
以聚酮合酶13为靶点的新型抗结核先导化合物的发现
2022-05-30 10:18:03王潇蒙建州关艳刘思含张佳炜李晓辉刘忆霜
中国抗生素杂志 2022年10期
王潇?蒙建州?关艳?刘思含?张佳炜?李晓辉?刘忆霜


摘要:目的 發现结核分枝杆菌Pks13(MtPks13)的抑制剂,为后续针对以MtPks13为靶点开发抗结核药物奠定了基础。方法 表达MtPks13-TE蛋白并优化其酶活测定方法,构建MtPks13抑制剂高通量筛选模型;与海分枝杆菌表型筛选方法联用,对5万个化合物进行筛选,将获得的抑制剂IMB-7142进行IC50以及酶动力学性质的测定;表面等离子共振(surface plasmon resonance, SPR)实验以及分子对接模型研究阳性化合物IMB-7142与MtPks13-TE蛋白是否能发生相互作用及其作用位点;用菌液稀释法评价IMB-7142对结核标准株的抗结核活性。结果 成功构建了MtPks13抑制剂筛选模型,并发现了一个对MtPks13有抑制作用,同时抑制海分枝杆菌生长的化合物IMB-7142,该抑制剂能与MtPks13-TE蛋白发生相互作用,具有较好的体外抗结核活性。结论 成功构建了稳定的MtPks13抑制剂高通量筛选模型,并且应用该模型筛选得到的一个抑制剂同时具有抗结核活性,为后续开发以MtPks13为靶点抗结核药物提供了思路。
关键词:结核分枝杆菌;Pks13;抑制剂;抗结核药物
中图分类号:R978.1文献标志码:A
Identification of novel anti-tuberculosis lead compound targeting
polyketide synthase 13
Wang Xiao1, Meng Jian-zhou2, Guan Yan1, Liu Si-han1, Zhang Jia-wei3, Li Xiao-hui1, and Liu Yi-shuang1
(1 National Laboratory for Screening New Microbial Drugs, Institute of Medicinal Biotechnology, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100050; 2 Shanghai Key Laboratory of Tuberculosis, Shanghai Pulmonary Hospital, Tongji University School of Medicine, Shanghai 200433; 3 School of Basic Medical Science, Jiamusi University, Jiamusi 154007)
Abstract Objective To discover inhibitors targeting Mycobacterium tuberculosis Pks13, and provide a foundation for the development of novel anti-tuberculosis drugs. Methods Mycobacterium tuberculosis Pks13-TE protein was overexpressed, and then the purified MtPks13-TE was used to establish a high-throughput screening model. In combination with the phenotype screening method of marine Mycobacterium, 50,000 compounds were screened, and the IC50 and enzyme kinetic properties of the lead candidate inhibitor, IMB-7142, were determined. Surface plasmon resonance (SPR) experiment and molecular docking model were used to study whether the positive compound IMB-7142 could interact with MtPks13-TE protein and, if so, its interaction sites. The anti-tuberculosis activity was evaluated by the broth dilution method. Results The screening model for MtPks13 inhibitors was constructed, and the compound IMB-7142 was found to inhibit MtPks13 and the growth of marine Mycobacterium. IMB-7142 could interact with MtPks13-TE and had anti-tuberculosis activity. Conclusion A stable high-throughput screening model for MtPks13 inhibitors was constructed, and the inhibitor obtained using this screening model exhibited anti-tuberculosis activity, which provided a starting point for the subsequent development of anti-tuberculosis drugs targeting MtPks13.
Key words Mycobacterium tuberculosis; Pks13; Inhibitor; Anti-tuberculosis drugs
结核病是由结核分枝杆菌(Mycobacterium tuberculosis,MTB)感染引起的一种严重危害人类健康的慢性传染病[1]。目前,它仍然是全球10大致死疾病之一。据世界卫生组织估算,2020年全球约有990万人患结核病,其中130万人因该病死亡,我国在30个结核病高负担国家中发病数位居第二[2]。2015年世界卫生组织(WHO)通过了1项终止全球结核病的战略决议,其目标是在2015—2030年之间将结核病发病率降低80%,死亡人数减少90%。然而,由于治疗方案不合理,患者依从性差,二线抗结核药物的使用不当以及代谢和营养差异等诸多因素,导致近年来多药耐药结核分枝杆菌(MDR-MTB)和广泛耐药结核分枝杆菌(XDR-MTB)的出现与快速传播。
从最新的统计数据可以看出,结核病治疗以及预防方面的全球目标和现实状况之间还存在着巨大差距。并且,近几十年来,仅有3种抗结核药物被批准上市。贝达喹啉(bedaquiline)于2012年获得美国FDA的批准用于结核病治疗,通过靶向ATP合成酶抑制所有敏感和耐药性MTB菌株[3-4],随后由日本大冢研发的德拉马尼(delamanid)获得欧洲FDA批准[5],其主要通过抑制MTB细胞壁霉菌酸的合成发挥作用[6],对耐药结核分枝杆菌表现出较强的抗菌活性。2019年美国FDA批准了TB联盟的pretomanid与贝达喹啉、利奈唑胺联用,治疗广泛耐药、不耐受或无反应的耐多药结核病。这3个上市药物正是因为其新的作用机制和靶点,使其能够有效地治疗MDR-MTB和XDR-MTB的感染。因此,發现抗结核药物新靶标,开发新型抗结核药物十分迫切。
分枝杆菌的细胞壁在分枝杆菌的生存力中起重要作用,因而其细胞壁是结核病药物开发新靶点的丰富来源。分枝菌酸则是构成结核分枝杆菌细胞壁的重要成分,它能形成渗透屏障,防止许多环境溶质进入,并赋予对许多抗生素的天然抗性。抑制分枝菌酸的生物合成已经被广泛认为是抗结核药物发现的一个高效策略[7-9]。在结核分枝杆菌中,聚酮合酶13(Pks13)主要负责分枝菌酸生物合成的最后一步,即C26 α-烷基支链和C40-60分枝菌酸脂前体的克莱森型缩合,最终生成α-烷基β-酮酸,而α-烷基β-酮酸是合成分枝菌酸的前体[10-11]。因此,Pks13在分枝菌酸的生物合成中起着至关重要的作用,同时它也是抑制这一途径的一个有效靶点。Pks13蛋白由5个结构域构成,包括:酮基合成酶结构域(keto synthase domain, KS)、酰基转移酶结构域(acyltransferase domain, AT)、两个酰基载体蛋白结构域(acyl-carrier protein domain, ACP) 和硫酯酶结构域(thioesterase domain, TE)。
其中TE结构域主要负责催化聚酮类化合物达到全长时从酶中的释放,这是形成分枝菌酸前体所必需的。2013年Loerger等[10]发现了一种对MTB H37Rv有活性的小分子TAM1,并确定其靶点为Pks13,后对抗性突变体进行全基因测序,发现突变位点位于Pks13的TE结构域。这提示Pks13的TE结构域被赋予作为结核药物靶点的巨大潜力。
本研究的目的是构建MtPks13-TE抑制剂高通量筛选模型,并应用此模型筛选获得MtPks13抑制剂,为寻找具有MtPks13抑制作用的结构新颖的抗结核先导化合物奠定基础。
1 材料与仪器
1.1 菌株、细胞株与载体
大肠埃希菌E. coli DH5α和E. coli BL21 (DE3) 为本室保存;海分枝杆菌(Mycobacterium marinum, BAA-535)由本室保存;结核分枝杆菌标准菌株H37Rv(ATCC27294)由北京胸科医院保存。人肝癌细胞HepG2和非洲绿猴肾细胞Vero由本室保存。pET28a-Pks13-TE质粒由本实验室前期构建并保存。
1.2 试剂与试剂盒
卡那霉素、异丙基硫代半乳糖苷(isopropyl thiogalactoside , IPTG)、Tris-HCl购自Amresco公司;CCK-8试剂盒购自Topscience公司,CCK-8可被活细胞中的脱氢酶还原成黄色甲瓒,可利用此特性进行细胞毒性检测;4-MUH购自Sigma公司;7H9液体培养基购自BD公司;质粒小提试剂盒购自Qiagen公司;蛋白浓度测定试剂盒购自Thermo公司。筛选所用化合物库来自国家新药(微生物)筛选实验室。
1.3 仪器
?KTA蛋白层析系统、His trapTM HP亲和层析柱为Cytiva公司产品;表面等离子共振(surface plasmon resonance, SPR)仪器为Richert公司;酶标仪为美国PerkinElmer公司的PerkinElmer Enspire 2300 Multilabel Reader型。
2 实验方法
2.1 结核分枝杆菌Pks13-TE蛋白的克隆、表达与纯化
结核分枝杆菌Pks13-TE蛋白的克隆、表达与纯化见之前的方法所述[12],最终将得到的成分中较纯的蛋白进行定量,加入甘油,冻于-80℃备用。
2.2 Pks13-TE酶活性的测定
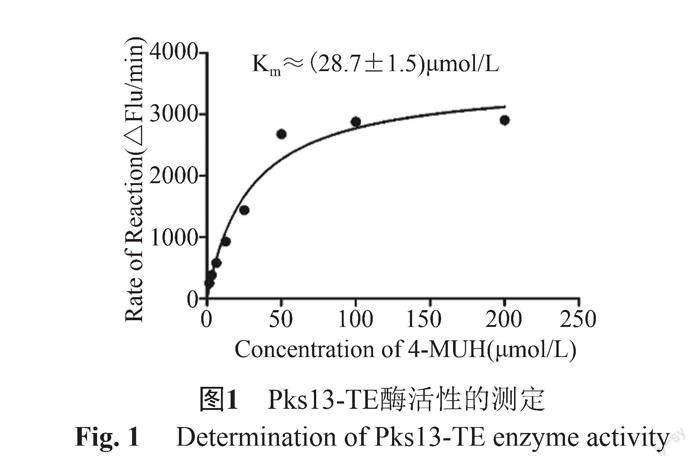
本研究使用4-甲基伞形基庚酸酯(4-methylumbelliferyl heptanoate,4-MUH)作为底物[10],在Pks13-TE蛋白的催化下,4-MUH会被水解为4-甲基伞形酮(4-methylumbelliferone,4-MU),通过在激发光355 nm、发射光460 nm处检测荧光值,来判断水解产物4-MU的量,从而反映出Pks13-TE蛋白的活性。
为了确定合适的底物浓度,将0.5 μmol/L Pks13-TE蛋白与不同浓度的4-MUH(1.56~200 μmol/L)加入96孔黑板(Costar)进行孵育,置于酶标仪37℃条件下检测荧光读数,5 min读1次,共检测120 min。使用前期处于线性范围内的点计算酶反应初始速率,以4-MUH浓度为横坐标,初始反应速率为纵坐标,使用GraphPad软件进行米氏方程拟合分析,确定动力学参数Km值。
2.3 海分枝杆菌对化合物库进行初筛
由于MTB需要在三级生物实验室进行操作,因此本研究选择与MTB基因相似性高达95%以上的海分枝杆菌作为体外筛选的模式菌,对化合物库中5万个样品进行初步筛选。将海分枝杆菌用7H9培养基(含10% OADC,0.05%吐温80)培养,每孔加入200 μL接种有对数期海分枝杆菌的7H9培养液(终浓度A600≈0.1),化合物筛选的终浓度为50 μg/mL,30℃静置培养5~7 d,观察结果。将抑制海分枝杆菌生长的化合物挑选出来,建立为初筛阳性库。
2.4 Pks13-TE蛋白抑制剂高通量筛选模型的建立与筛选
运用2.2所述方法,对酶促反应时间和酶的用量进行优化。依据Km值,设置底物4-MUH濃度为30 μmol/L,然后与不同浓度的Pks13-TE蛋白(0~7.2 μmol/L)孵育,置于酶标仪37℃条件下检测荧光读数,10 min读一次,共检测120 min。考虑到高通量筛选模型信号背景比大于3,且孵育时间不宜过长、酶用量不宜过大以确保通量,最终确定的酶活检测体系如下:0.1 mol/L Tris-HCl pH 7.0,30 μmol/L 4-MUH,1.8 μmol/L Pks13-TE蛋白,反应时间为20 min。
运用上述优化的酶活测定方法,对建立的初筛阳性化合物库中样品以终浓度20 μg/mL进行筛选,同时设置阴性对照组(含相同含量的DMSO)和阳性对照组(不加入Pks13-TE蛋白,即酶活性完全被抑制)。抑制率计算公式如下:酶抑制率=(阴性对照组-加入化合物的实验组)/(阴性对照组-阳性对照组)×100%。经过初筛和复筛之后,得到抑制效果最好的抑制剂IMB-7142。
2.5 IMB-7142对海分枝杆菌的MIC测定
海分枝杆菌用7H9培养基(含10% OADC,0.05%吐温80)培养,在96孔板中。每孔加入200 μL接种有对数期海分枝杆菌的7H9培养液(终浓度A600≈0.1),化合物的终浓度依次为0、0.25、0.5、1、2、4、8、16、32、64和128 μg/mL,30℃静置培养5~7 d观察结果,孔内完全抑制细菌生长的最小药物浓度为最低抑菌浓度(minimum inhibitory concentration,MIC)。同时设置阴性对照组(含相同DMSO含量的7H9培养基)和阳性对照组(异烟肼 0.5 μg/mL)。
2.6 IMB-7142在Pks13-TE蛋白抑制剂模型上的IC50测定
将筛选得到的阳性化合物用DMSO配置成10 mg/ mL的母液,将化合物倍比稀释,使其终浓度分别为0.78、1.56、3.125、6.25、12.5、25、50和100 μg/mL,按照上述的酶活体系,分别获得化合物不同浓度条件下的抑制率,以化合物浓度的对数为横坐标,抑制率为纵坐标,利用Graphpad prism 5软件进行数据拟合分析,获得半数抑制浓度(half maximal inhibitory concentration,IC50)。
2.7 IMB-7142对Pks13-TE蛋白抑制作用的酶动力学研究
筛选条件下,分别将底物4-MUH、化合物IMB-1742进行浓度倍比稀释,对于同一浓度的化合物,设置7个4-MUH浓度梯度(120、60、30、15、7.5、3.75和1.875 μmol/L),同时对阳性化合物设置5个浓度梯度(100、50、25和12.5 μg/mL)。按照反应速率=(FT2- FT1)/(T2-T1)计算各底物浓度下对应的不同化合物浓度的反应速率。采用Lineweaver-Burk作图法以4-MUH浓度的倒数为横坐标,以反应速率的倒数为纵坐标,绘制不同抑制剂浓度作用下的曲线,依据这些直线的交点位置推测抑制剂的类型。
2.8 SPR实验检测IMB-7142与Pks13-TE蛋白的结合情况
SPR实验首先将Pks13-TE蛋白固定在带有葡聚糖的CM5芯片上,随后使用PBST缓冲液将阳性化合物梯度稀释(100、50和25 μmol/L),保证每个浓度均含5%的DMSO,后将稀释好的化合物由低浓度到高浓度依次流经芯片。SPR实验使用Reichert 2SPR system仪器完成,结果使用TraceDrawer软件进行分析。
2.9 分子对接分析IMB-7142与Pks13-TE蛋白的相互作用位点
为了探究阳性化合物的结构如何影响其对Pks13-TE蛋白的抑制作用,使用了Discovery Studio 2019对化合物和Pks13-TE进行分子对接分析。Pks13-TE的晶体结构来自于Protein Data Bank数据库(PDB:5V3Y),其蛋白结构由一个核心结构域和一个盖子结构域组成,活性口袋位于这两个结构域之间的界面。用分子对接软件对IMB-7142分子结构进行优化,并与活性口袋进行对接,选出打分最高的对接模型分析其相互作用。
2.10 IMB-7142抗结核活性的测定
将待测化合物用DMSO溶解后,在96孔板检测IMB-7142对Mtb H37Rv 的MIC,同时设置异烟肼为阳性对照。每孔加入200 μL接种有对数期Mtb H37Rv的7H9培养液(终浓度A600≈0.1),化合物的终浓度依次为0、0.25、0.5、1、2、4、8、16、32、64和128 μg/mL,37℃静置培养7 d观察结果,获得MIC。
2.11 CCK-8检测化合物的细胞毒性
HepG2细胞以5×104/mL接种于96孔细胞培养板,Vero细胞以2×104/mL接种于96孔细胞培养板,每孔体积100 μL,同时96孔细胞培养板的边缘孔以无菌PBS填充,尽量避免边缘效应。将细胞置于37℃,5% CO2条件下过夜培养,使细胞贴壁。使用含10%FBS的DMEM培养基将化合物IMB-7142进行2倍梯度稀释10个浓度(200~0.39 μmol/L),每孔加入200 μL。各个浓度均做3个复孔,并设置含等量DMSO的对照组和无细胞仅含培养基的空白组。培养48 h后,每孔加入20 μL的CCK-8,于细胞培养箱37℃孵育1~4 h,用酶标仪检测在450 nm下的吸光值,计算细胞存活率。存活率计算公式如下:存活率=(给药组A值-空白组A值)/(对照组A值-空白组A值)×100%。以化合物浓度的对数为横坐标,存活率为纵坐标,利用Graphpad prism 5软件进行数据拟合分析,获得IC50。
3 结果
3.1 Pks13-TE的活性测定
在Pks13-TE蛋白的催化下,底物4-MUH会被水解为4-MU,在激发光355 nm、发射光460 nm处检测荧光值,通过水解产物4-MU的变化量来反应Pks13-TE的催化活性。结果显示,随着4-MUH浓度的增加,Pks13-TE的活性越来越强,直至达到平台期,Km值为28.7 μmol/L(图1)。
3.2 海分枝杆菌BAA-535对化合物库进行初筛
利用模式菌海分枝杆菌对化合物库中的5万样品进行筛选,如果化合物能够完全抑制海分枝杆菌的生长,则被认为是初筛阳性的化合物。通过筛选,共得到849个初筛阳性的化合物,建立初筛阳性化合物库。
3.3 Pks13-TE抑制剂高通量筛选模型的建立与筛选
以实验方法2.2所述为基础,将蛋白进行梯度稀释,检测120 min(每隔10 min检测一次)内酶促反应情况,以对酶活反应体系中的酶用量和反应时间进行优化(图2A)。依据高通量筛选模型信号噪声比>3以及高通量筛选过程的时间成本,最终选定的酶活检测条件为蛋白用量1.8 μmol/L,反应时间为20 min。经评价,模型的Z'因子为0.98,信号噪音比、信号背景比均符合高通量筛选的要求。对初筛阳性化合物库中的849个化合物进行筛选,最终得到一个具有较好抑制活性的化合物IMB-7142,其结构如图2B所示。
3.4 阳性化合物IMB-1742对Pks13-TE抑制作用的IC50测定
IMB-1742对Pks13-TE活性抑制作用如图2C所示,随着化合物浓度的增高,对Pks13-TE蛋白抑制作用逐渐增强,表现出明显的浓度依赖性。通过Graphpad Prism 5软件对曲线进行拟合分析,可得阳性化合物IMB-1742对Pks13-TE酶活性的IC50约为14.52 μg/mL。
3.5 阳性化合物IMB-1742对 Pks13-TE抑制作用的酶动力学性质的研究
对IMB-1742抑制Pks13-TE活性的作用类型进行研究,在不同浓度的抑制剂作用下,反应速度随底物浓度而变化。采用Lineweaver-Burk曲线法,以底物浓度的倒数为横坐标,反应速率的倒数为纵坐标作图。结果如图3显示,不同浓度IMB-1742的抑制曲线交于纵轴正半轴,推测阳性化合物IMB-1742可能为4-MUH的竞争性抑制剂。
3.6 SPR实验检测阳性化合物IMB-1742与Pks13-TE蛋白的相互作用
SPR实验由Reichert 2SPR system仪器完成,将PBST缓冲液梯度稀释的阳性化合物(100、50和25 μmol/L)由低濃度到高浓度依次流经固定有Pks13-TE蛋白的CM5芯片,结果显示,IMB-1742能够剂量依赖性的与Pks13-TE蛋白结合(图4),其结合的KD值为4.08×10-4 mol/L。
3.7 阳性化合物IMB-1742与Pks13-TE的分子对接结果
Pks13-TE蛋白的活性中心位于其核心结构域和盖子结构域之间的界面,将配体结合到Pks13-TE蛋白的活性中心,结果如图5所示,IMB-1742结构中的酰胺基团的活泼氢原子作为氢键供体能够与Asn1640形成氢键作用力。噻吩取代基可以与Tyr1663、Tyr1674形成π-π堆积相互作用,结构中大的芳香母核结构与His1664同样形成π-π堆积相互作用。同时,四氢吡咯中的氮原子与Asp1644和Phe1670两个氨基酸残基分别存在Pi-Cation相互作用力。此外,IMB-1742还能与Ser1533等形成范德华力。
3.8 阳性化合物IMB-1742的抗菌活性评价
阳性化合物IMB-1742对海分枝杆菌、结核分枝杆菌标准株H37Rv的MIC分别为16和8 μg/mL(表1)。
3.9 阳性化合物IMB-1742的细胞毒性检测结果
采用CCK-8试剂盒检测IMB-1742对HepG2和Vero细胞的毒性影响,结果如图6所示,阳性化合物对HepG2细胞生长抑制的IC50为73.61 μmol/L,对Vero细胞生长抑制的IC50为79.94 μmol/L,说明IMB-1742对细胞毒性较小。
4 讨论
分枝菌酸是分枝杆菌细胞壁中含量丰富且特异的脂质成分[13]。通过切断合成分枝菌酸的通路,除可产生抗结核作用外,还可提高其他抗结核药物在菌体的浓度,利于组建联合用药方案[14]。因此,分枝菌酸生物合成途径中的各种酶可作为结核病药物研究的靶点来源。Pks13是分枝菌酸生物合成最后一步的关键蛋白,它对分枝杆菌的生存至关重要[15]。如果Pks13蛋白被抑制,分枝菌酸合成途径受阻,将会破坏结核分枝杆菌的细胞壁,最终导致菌体的死亡。因此,以MtPks13蛋白为靶点,可能会获得具有抗结核活性的先导化合物。近年来,仅有4类Pks13抑制剂被报道,分别是噻吩类[16]、苯并呋喃类[10]、香豆雌酚类[17-18]和β-内脂类[19],目前对具有新型母核结构、且具有抗结核活性的Pks13抑制剂有着强烈的需求。
本研究对结核分枝杆菌Pks13蛋白的TE结构域进行了表达纯化,通过酶活性的测定与评价,建立了以MtPks13-TE为靶点的高通量筛选模型。利用该模型对初筛得到的849个具有抗海分枝杆菌活性的化合物进行再次筛选,选取活性较好的IMB-1742进行进一步的研究。IMB-1742虽然包含苯并呋喃环,但此环连接了一个柔性长链基团,这点不同于已发现的苯并呋喃类Pks13抑制剂。我们通过SPR实验证明IMB-1742能够与MtPks13-TE蛋白发生相互作用,其KD值为4.08×10-4 mol/L,属于中等强度的结合。进一步,我们使用分子对接软件分析了化合物如何抑制MtPks13-TE的活性。MtPks13-TE活性中心位于核心结构域和盖子结构域之间的界面,结果显示,IMB-1742能与MtPks13-TE蛋白的多个活性关键氨基酸(Asn1640、Phe1670、Asp1644、Ser1533)形成相互作用,說明化合物很可能与底物竞争性结合蛋白的活性位点,进而抑制蛋白的催化功能,从而发挥抗菌活性。SPR和分子对接实验为后续修饰化合物IMB-1742提供了方向,以增强与蛋白结合的亲和力和稳定性,提高抗菌活性。
本研究获得了1个MtPks13-TE的选择性抑制剂IMB-1742。该化合物表现出较好的体外抗结核活性, 可与MtPks13-TE蛋白结合并能浓度依赖性的抑制其活性。IMB-1742没有表现出明显的细胞毒性, 通过结构改造有可能成为抗结核先导化合物,为针对以MtPks13为靶点开发抗结核药物提供了新思路。
参 考 文 献
Mishra, R, Shukla, P, Huang, W, et al. Gene mutations in Mycobacterium tuberculosis: Multidrug-resistant TB as an emerging global public health crisis[J]. Tuberculosis, 2015, 95(1): 1-5.
WHO Global tuberculosis report. 2021.
Diacon A H, Pym A, Grobusch M, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis[J]. N Engl J Med, 2009, 360(23): 2397-2405.
Lakshmanan M, Xavier A S. Bedaquiline-the first ATP synthase inhibitor against multi drug resistant tuberculosis[J]. J Young Pharm, 2013, 5(4): 112-115.
Blair H A, Scott L J. Delamanid: A review of its use in patients with multidrug-resistant tuberculosis[J]. Drugs, 2015, 75(1): 91-100.
Matsumoto M, Hashizume H, Tomishige T, et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice[J]. PLoS Med, 2006, 3(11): e466.
Abrahams K A, Besra G S. Mycobacterial cell wall biosynthesis: A multifaceted antibiotic target[J]. Parasitology, 2018, 145(2): 116-133.
Takayama K, Wang C, Besra G S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis[J]. Clin Microbiol Rev, 2005, 18(1): 81-101.
吕润秋, 张维, 于丽芳. 靶向分枝菌酸生物合成的抗结核化合物研究进展[J]. 有机化学, 2021, 41(6): 2249-2260.
Aggarwal A, Parai M K, Shetty N, et al. Development of a novel lead that targets M. tuberculosis polyketide synthase 13[J]. Cell, 2017, 170(2): 249-259.e225.
Lun S, Xiao S, Zhang W, et al. Therapeutic potential of coumestan Pks13 inhibitors for tuberculosis[J]. Antimicrob Agents Chemother, 2021, 65(5): e02190-20.
Wang X, Zhao W, Wang B, et al. Identification of inhibitors targeting polyketide synthase 13 of Mycobacterium tuberculosis as antituberculosis drug leads[J]. Bioorg Chem, 2021, 114: 105110.
Veyron-Churlet, R, Bigot, S, Guerrini, O, et al. The biosynthesis of mycolic acids in Mycobacterium tuberculosis relies on multiple specialized elongation complexes interconnected by specific protein-protein interactions[J]. J Mol Biol, 2005, 353(4): 847-858.
丁威, 赵文婷, 张东峰. 结核分枝杆菌聚酮合成酶13抑制剂的研究进展[J]. 药学学报, 2020, 55(8): 1768-1773.
Portevin D, De Sousa-DAuria C, Houssin C, et al. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms[J]. Proc Natl Acad Sci U S A, 2004, 101(1): 314-319.
Thanna S, Knudson S E, Grzegorzewicz A, et al. Synthesis and evaluation of new 2-aminothiophenes against Mycobacterium tuberculosis[J]. Org Biomol Chem, 2016, 14(25): 6119-6133.
Zhang W, Lun S, Wang S H, et al. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis[J]. J Med Chem, 2018, 61(3): 791-803.
Zhang W, Lun S, Liu L L, et al. Identification of novel coumestan derivatives as polyketide synthase 13 inhibitors against Mycobacterium tuberculosis. Part II[J]. J Med Chem, 2019, 62(7): 3575-3589.
Lehmann J, Cheng T Y, Aggarwal A, et al. An antibacterial β-lactone kills Mycobacterium tuberculosis by disrupting mycolic acid biosynthesis[J]. Angew Chem Int Ed Engl, 2018, 57(1): 348-353.
收稿日期:2022-04-13
作者簡介:王潇,女,生于1988年,助理研究员,博士,主要从事抗结核药物的发现和机制研究,E-mail: wangxiao_1107@163.com
*通讯作者,E-mail: liuys@imb.pumc.edu.cn
猜你喜欢
天然产物研究与开发(2018年7期)2018-08-21 02:03:54
上海农业学报(2017年3期)2017-04-10 12:39:26
中国实用医药(2016年18期)2016-08-03 09:49:34
中国实用医药(2016年9期)2016-05-17 10:59:06
中国民族医药杂志(2016年2期)2016-05-14 07:12:05
中外医疗(2015年3期)2015-08-29 20:31:50
医学信息(2015年14期)2015-04-29 17:36:37
中国社区医师(2014年22期)2015-04-16 21:44:21
中国当代医药(2015年20期)2015-03-01 02:04:25
中国民族民间医药·下半月(2014年6期)2015-01-21 06:24:29
