
蛋白质序列比对下的分子虚拟筛选方法
2022-05-27 01:06郑琳琳贾宁欣张贵军
小型微型计算机系统 2022年6期
郑琳琳,贾宁欣,张贵军,胡 俊
(浙江工业大学 信息工程学院,杭州 310023)
1 引 言
识别与受体蛋白质进行相互作用并适当修改其生物学行为的活性分子是药学研究面临的一项基本挑战.据统计,一种新药物从筛选到成功上市,平均需要10年~14年的时间,中间过程的花费高达2亿~3.5亿美元,其中,候选药物分子在临床实验阶段的淘汰率高达90%[1].因此,如何快速高效地筛选出有效的候选药物分子,提高临床实验阶段的成功率成为了各大制药公司和学术机构的研究重点[2].传统的高通量筛选技术以分子水平和细胞水平的实验方法为基础,存在筛选结果中假阳性的比例较高和小分子的非特异性聚集等问题,同时高通量筛选技术所使用的试剂和仪器都非常昂贵,仅有一些大型制药公司和少数学术机构能负担得起[3].随着数字化信息时代的到来,越来越多的生物信息以数字化的方式得以保存,因此利用计算机进行药物虚拟筛选受到越来越多研究者的关注[4].
药物虚拟筛选是指进行生物活性筛选之前,利用生物计算手段对待筛选的药物分子库进行初步筛选,降低生物实验筛选化合物的数目,从而缩短药物筛选的周期[5].已有的药物虚拟筛选方法可以主要分为两类:基于需要事先给定一个或一组能与受体蛋白质相互作用的活性分子.根据给定的活性分子与待筛选分子间的相似性,基于活性分子的方法将待筛选的所有分子进行排序,再根据目标筛选数目进行筛选结果的返回.由于基于活性分子的方法需要事先给定能与受体蛋白质相互作用的活性分子信息,使得该类方法很难得到推广.基于蛋白质结构的方法[6,7]需要事先给定高分辨率的受体蛋白质结构,其筛选性能很大程度上取决于给定受体蛋白质结构的质量和灵活性[8].基于蛋白质结构的方法进一步地又可分为基于分子对接与基于结构相似性的方法.其中,基于分子对接方法主要步骤为:首先,使用分子对接工具(如:AutoDock Vina[9])将待筛选分子与受体蛋白质结构进行对接,然后,根据每个待筛选分子的对接得分将它们进行排序,最后,根据目标筛选数目进行筛选结果的返回;基于结构相似性的方法[10]主要步骤为:首先,根据受体蛋白质结构信息从蛋白质与活性分子相互作用库(如:Protein Data Bank,PDB[11])进行结构相似性搜索,将搜索出来的活性分子当成能与受体蛋白质发生相互作用的潜在活性分子,然后,根据这些潜在活性分子,使用基于活性分子方法对待筛选分子进行筛选.
基于分子对接的方法需要将每个待筛选分子与受体蛋白质结构进行对接,极其耗时费力.例如:基于AutoDock Vina工具的虚拟筛选方法,将每个分子对接到受体蛋白结构中需要花费1.16分钟[9].为了方便描述,下文我们将基于AutoDock Vina实现的基于分子对接的虚拟筛选方法亦称之为AutoDock Vina.相比之下,基于结构相似性的方法就体现出省时的优势,仅在搜索潜在活性分子阶段花费一定时间,在分子相似性评估阶段花费的时间远远少于基于分子对接的方法.常见的基于结构相似性的方法有:FINDSITEX[12]、FINDSITEfilt[13]、FINDSITEcomb[13]、PoLi[14]、SPOT-Ligand[15]和SPOT-Ligand 2[10].FINDSITEX和FINDSITEfilt都基于穿线识别算法,具有与基于活性分子的方法一样快的优势,并且克服了基于活性分子的方法无法对活性分子未知的受体蛋白质进行药物筛选的问题.上述两种方法可以利用预测的受体蛋白质的低分辨率结构,根据结构相似性指标,从蛋白质与分子相互作用数据库中获取的潜在活性分子信息来推断活性分子与受体蛋白质相互作用的可能性.FINDSITEcomb方法则是将FINDSITEX和FINDSITEfilt结合起来.PoLi是一种基于结构相似性的方法,它集成了基于2D指纹和3D结构的相似性度量,实现分子虚拟筛选功能.SPOT-Ligand亦是一种基于结构相似性的方法,它通过计算两两分子之间的Tanimoto系数[16]来评价分子2D指纹之间的相似性,通过使用SPalign[17]来计算两两分子之间的3D结构之间的相似性.SPOT-Ligand 2是SPOT-Ligand的扩展版,它的模板库相较于SPOT-Ligand的扩展了15倍,从而进一步地提高了虚拟筛选性能.
虽然基于结构相似性的方法在分子虚拟筛选性能上具有一定的优势,但是他们通常需要受体蛋白质的结构信息,而对于大多数蛋白质来说,结构信息是未知的.虽然可以通过生物计算方法[18-21]来预测受体蛋白质的结构,但是预测过程需要花费较长时间且预测得到的结构的质量会对分子虚拟筛选结果产生巨大的影响.因此,本文从受体蛋白质的序列出发,提出了一种名为Screener的蛋白质序列比对和活性分子相似性评估的虚拟筛选方法.对任一待筛选活性分子的受体蛋白质序列,Screener首先生成其位置特异性频率矩阵(PSFM)特征、预测的二级结构(PSS)特征以及预测的溶剂可及性(PSA)特征,并利用一种基于蛋白质序列的结合位点残基预测方法I-LBR[22]来预测受体蛋白质与分子的结合位点残基;其次,根据预测的结合位点残基以及其对应特征来搜索BioLiP[23]、DrugBank[24]和BindingDB[25]这3个蛋白质与分子相互作用的数据库,构建待筛选蛋白质特异性的模板蛋白质库;然后,将所有与任意模板蛋白质相互作用的活性分子收集起来构建潜在的种子分子库;最后,利用分子2D指纹之间的相似性来对待筛选分子集进行排序,筛选出受体蛋白质的活性分子.在基准测试集DUD40和DUD-E65上的实验结果验证了Screener的有效性.
2 Screener算法设计
2.1 数据集
本文利用数据集DUD[26]和DUD-E[27]来验证Screener的虚拟筛选性能.DUD数据集包含40个受体蛋白,每个受体蛋白的活性分子和诱饵分子的比例为1:36,命名为DUD40.DUD-E数据集包含102个受体蛋白,每个受体蛋白平均有224个活性分子,平均每个活性分子对应于50个诱饵分子.受PoLi[14]、SPOT-Ligand[15]和SPOT-Ligand 2[10]的启发,本文中去除了已经包含在DUD40数据集中的37个受体蛋白质,剩下的65个受体蛋白质组成一个新的数据集,命名为DUD-E65.
2.2 特征表示
如何抽取蛋白质的特征信息一直受到生物信息学领域的重点关注.特征表示的优劣将直接影响蛋白质相关属性的预测性能的好坏.常见的蛋白质特征表示方法有氨基酸组成成分、位置特异性频率矩阵信息与二级结构信息等.为了充分利用蛋白质信息提升分子虚拟筛选的性能,本文使用了位置特异性频率矩阵(PSFM)特征、预测的二级结构(PSS)特征以及预测的溶剂可及性(PSA)特征来表示受体蛋白质的特征信息.
PSFM特征可以反映出蛋白质的序列信息和进化信息,通过利用PSI-BLAST[28]工具搜索非冗余序列数据库NCBI[29]生成多序列联配文件(Multiple Sequence Alignment,MSA),然后根据MSA信息,通过公式(1)生成大小为L×20的PSFM特征:
(1)
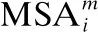
PSS指的是多个残基在局部所表现出的一种链接状态,这里表示为一个大小为L×3的预测二级结构矩阵,它是利用PSIPRED[30]程序在默认参数下生成的.在PSS中,每行表示相应残基的3种二级结构类别(无规卷曲(C)、α-螺旋(H)和β-折叠(E))的概率.
PSA指蛋白质每个残基可以与水接触的面积大小,间接反映了这个残基是否可溶于水,这里表示为一个大小为L×3的预测溶剂可及性矩阵,它是利用SANN[31]程序在默认参数下生成的,其中每行表示相应残基的3种溶剂可及性状态(掩埋(B)、中间(I)和暴露(E))的概率.
2.3 特异性结合位点残基预测
本文使用I-LBR[22]对受体蛋白质进行特异性结合位点残基预测.I-LBR是一种从蛋白质序列出发,通过特异性查询的计算方法进行蛋白质结合位点残基的预测.I-LBR首先收集与结合位点相关的残基的模板数据库,它利用PSI-BLAST[28]、PSIPRED[30]和SANN[31]程序生成BioLiP[23]数据库中每条蛋白质的位置特异性得分矩阵(Position-Specific Scoring Matrix,PSSM)、PSS和PSA特征,通过比对受体蛋白质与BioLiP中所有序列相似性小于30%的蛋白质的上述3个特征信息,来生成受体蛋白质特异性的训练子集;然后使用支持向量机[32]算法训练受体蛋白质特异性的预测模型;最后,利用受体蛋白质特异性的预测模型对受体蛋白质进行结合位点残基预测.
2.4 Screener
受I-LBR[22]的启发,本文提出了一种名为Screener的基于蛋白质序列比对和活性分子相似性评估的分子虚拟筛选方法.对任一待筛选活性分子的受体蛋白质序列,Screener首先生成其位置特异性频率矩阵(PSFM)特征、预测的二级结构(PSS)特征以及预测的溶剂可及性(PSA)特征,并利用一种基于蛋白质序列的结合位点残基预测方法I-LBR[22]来预测受体蛋白质与分子的结合位点残基;其次,根据预测的结合位点残基以及其对应特征来搜索BioLiP[23]、DrugBank[24]和BindingDB[25]3个蛋白质与分子相互作用的数据库,构建待筛选蛋白质特异性的模板蛋白质库;然后,将所有与任意模板蛋白质相互作用的活性分子收集起来构建潜在的种子分子库;最后,利用分子2D指纹之间的相似性来对待筛选分子集进行排序,筛选出受体蛋白质的活性分子.图1显示了Screener的示意图.
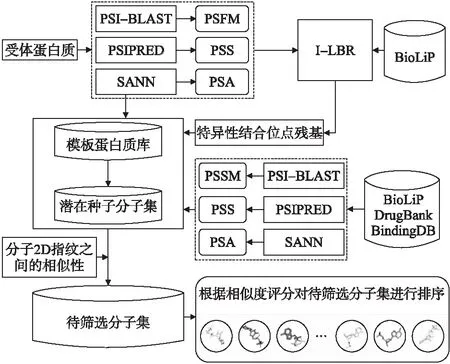
图1 Screener框架图
2.4.1 寻找模板蛋白质库和潜在的种子分子集
本文使用I-LBR预测受体蛋白质与分子的结合位点残基,根据预测的结合位点残基信息及PSFM、PSS和PSA特征信息,从BioLiP[23]、DrugBank[24]和BindingDB[25]蛋白质与分子相互作用的数据库中搜索相似蛋白质,来构成模板蛋白质库.将所有与任意模板蛋白质相互作用的活性分子收集起来构建潜在的种子分子库.具体来说,对BioLiP[23]、DrugBank[24]和BindingDB[25]中的任意一个模板蛋白质,本文首先使用PSI-BLAST[28]、PSIPRED[30]和SANN[31]程序来生成对应的PSSM、PSS以及PSA特征;其次,使用I-LBR预测受体蛋白质中的分子结合位点残基;再使用公式(2)计算受体蛋白质(记作q)与上述3个数据库中的任一模板蛋白质(记作t)之间的相似矩阵S:
(2)
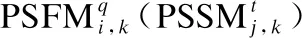
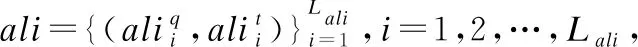
(3)
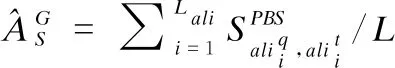
从BioLiP、DrugBank和BindingDB这3个蛋白质与分子相互作用的数据库中,选择那些与受体蛋白质的对应Q大于0.5且序列一致性小于30%的蛋白质组成模板蛋白质库.如果选择到的模板蛋白质条数少于5条,则选择Q得分高且与受体蛋白质序列一致性小于30%的前5条蛋白质组成模板蛋白质库.然后,将与模板蛋白质库中任一蛋白质相互作用的活性分子收集起来组成潜在的种子分子集.
2.4.2 对待筛选分子集进行排序
为了充分利用上述收集到的潜在种子分子集的信息,本文首先使用分子2D指纹相似性作为度量,来对种子分子进行聚类.具体而言,本文使用分子2D指纹之间的Tanimoto系数(记作TC)来作为分子间相似性度量,使用简单的链接聚类算法进行聚类,包含以下步骤:1)每个种子分子被视为一个簇;2)当两个簇的TC值高于0.9时,将被合并为一个簇;3)合并满足条件的簇,直至没有需要合并的簇为止.这里,两个簇之间的TC值定义为所有来自不同簇的两个分子之间的TC值中的最大值;两个分子之间的TC值计算方式如公式(4)所示:
(4)
其中,a为第1个分子2D指纹中的值为1的比特位元素数目,b为第2个分子2D指纹中的值为1的比特位元素数目,c为两个分子2D指纹中的对应位置值均为1的比特位元素数目.本文使用OpenBabel[36]软件来生成每个分子的1024比特位2D指纹信息.
为了充分使用每个类簇中的分子信息,进而提升虚拟筛选性能,本文设计了一种新的相似度评分方式来计算任一待筛选分子与受体蛋白质相互作用的可能性:
(5)
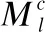
2.5 评价指标
在虚拟筛选领域,受试者工作特性(ROC)[37]曲线、筛选数据库的富集因子(EF)和命中率(HR)3个评价指标经常被用来衡量筛选性能的好坏.具体来说,ROC曲线表示真阳性率与假阳性率之间的函数.通过计算ROC曲线下方与坐标轴围成的面积(记作AUC)来量化ROC曲线的优劣,AUC值属于0~1之间,小于0.5表示该方法仅实现了随机性能;EF和HR是两个重要的评价指标,用于评价待筛选分子集中排名前x%数据的性能.因此,本文亦使用AUC、EF和HR来衡量Screener的筛选性能.
这里,EF的定义为:
(6)
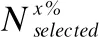
(7)
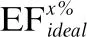
3 实验结果与分析
3.1 I-LBR有助于提升分子虚拟筛选的性能
为了评估I-LBR预测的结合位点残基信息是否有助于提升分子虚拟筛选的性能,本文在不利用I-LBR预测的结合位点残基信息的基础上,实现了另一种分子虚拟筛选方法,命名为weakScreener.这里,weakScreener和Screener的其余步骤完全一致.本文在数据集DUD40和DUD-E65上将weakScreener与Screener进行比较.
表1展示了Screener和weakScreener在数据集DUD40和DUD-E65上获得的平均EF和HR值.在数据集DUD40上,Screener的EF1%、EF5%、HR1%和HR5%的平均值分别为16.6、9.3、44.1和46.1,均高于weakScreener(EF1%=15.5、EF5%=9.1、HR1%=41.2和HR5%=45.4).尽管Screener在DUD40上的HR10%值稍低于weakScreener,但是在DUD-E65上,Screener的EF1%、EF5%、EF10%、HR1%、HR5%、HR10%的平均值分别为25.7、10.9、6.1、67.6、55.0、60.9,分别比weakScreener高出14.7%、21.1%、17.3%、16.6%、22.5%、17.6%.总的来说,由表1我们可以得出Screener具有比weakScreener更好的分子虚拟筛选性能,同时也证明了,通过I-LBR预测的受体蛋白质的结合位点残基有助于提升筛选性能的提升.
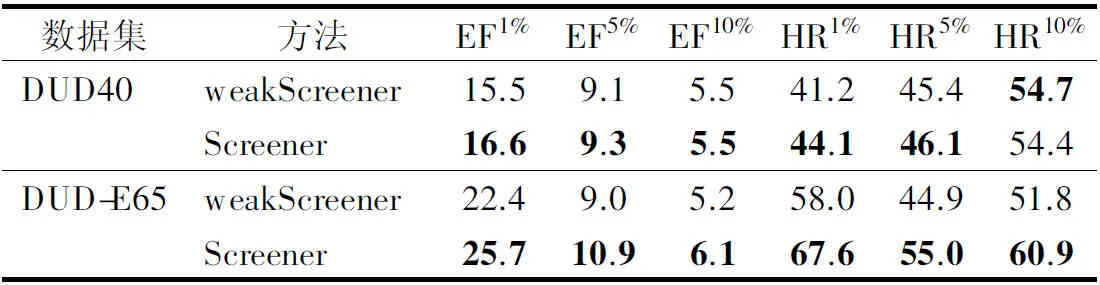
表1 Screener和weakScreener在DUD40和DUD-E65数据集上的平均EF和HR的比较
3.2 与现有分子虚拟筛选方法的比较
表2列出了Screener和AutoDock Vina[9]、FINDSITEfilt[13]、PoLi[14]等现有方法在数据集DUD40和DUD-E65上的筛选性能.值得注意的是,除Screener以外其他方法均需要使用蛋白质的结构信息,表2中列出的其他方法使用了TASSER-VMT[38]建模的结构信息.在数据集DUD-E65上,Screener、PoLi、FINDSITEfilt和AutoDock Vina实现的平均EF1%分别为25.7、9.6、7.9和3.0.在DUD40数据集上也观察到类似的趋势,Screener、PoLi、FINDSITEfilt和AutoDock Vina的平均EF1%分别达到16.6、13.4、9.0和1.6.
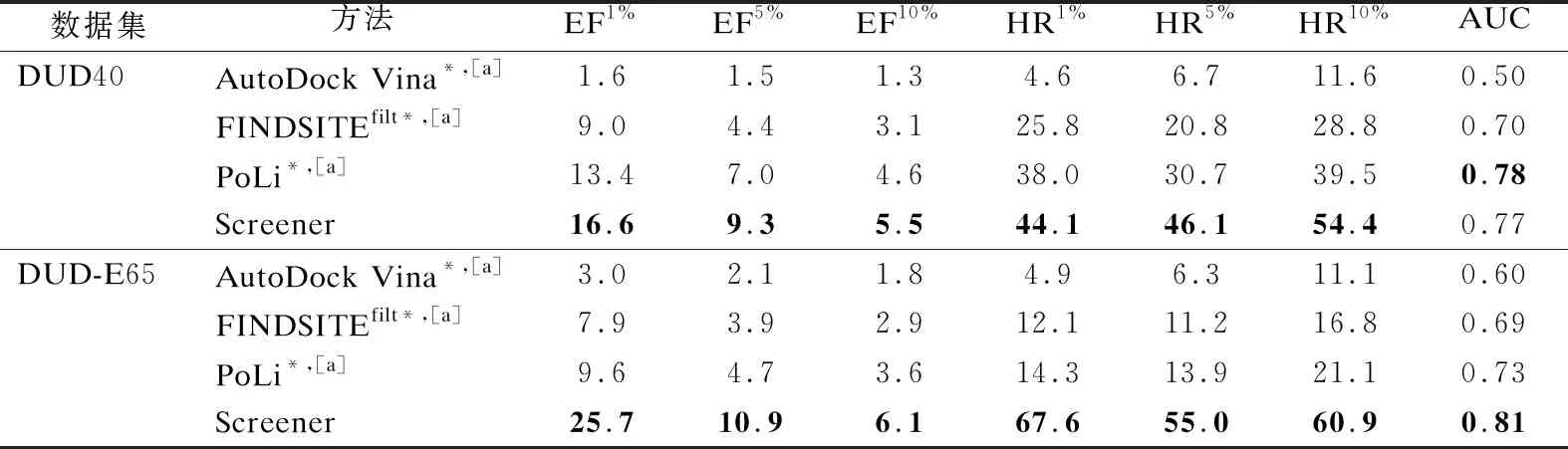
表2 Screener与基于建模结构的其他虚拟筛选方法在DUD40和DUD-E65数据集上的性能比较
图2给出了Screener和PoLi在数据集DUD40和DUD-E65上EF1%值的比较,图2中每个面板上的数字分别代表上三角形和下三角形上的点数.PCC为Screener与PoLi的EF1%值之间的皮尔森相关系数.p-value值由Student′s t检验计算得到.由图2可以发现,Screener在两个数据集上都优于PoLi.以DUD-E65为例(图2(b)),在65个试验受体蛋白质中,其中有57个受体蛋白质通过Screener筛选的的EF1%高于PoLi.此外,使用Screener得到的EF1%>30的受体蛋白质数量为32个,明显高于使用PoLi得到EF1%>30的受体蛋白质数目(4个).值得一提的是,Screener在两个受体蛋白质(HXK4和THB)上筛选失效,未能获得富集因子,即EF1%=0;相比之下,PoLi在8个受体蛋白质(CP2C9,CXCR4,HIVINT,HXK4,ITAL,PYRD,THB,TRYB1)上筛选失效.在Student′st检验中,Screener和PoLi的EF1%差值具有统计学意义,它们的p-value小于10-9.表3进一步展示了Screener和基于生物实验测定的蛋白质结构的其他虚拟筛选方法(如:AutoDock Vina[9]、FINDSITEcomb[13]、PoLi[14]、SPOT-Ligand[15]和SPOT-Ligand 2[10])在数据集DUD40和DUD-E65上的平均EF性能的比较.
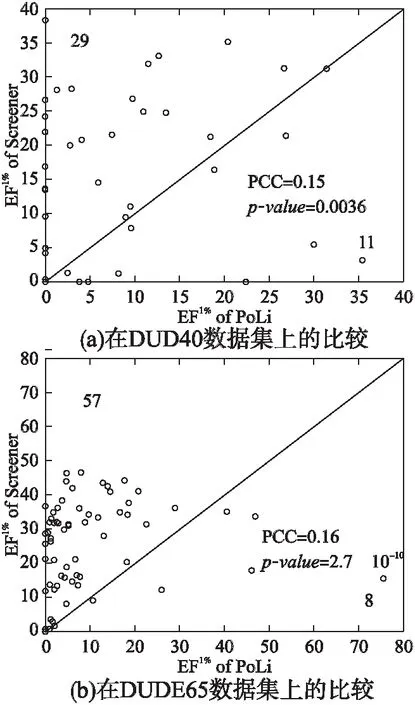
图2 Screener和PoLi的EF1%值比较
结果节选自[a][14],*这些方法需要受体蛋白的结构.
由表3可知,在DUD40数据集上,Screener的EF1%、EF5%和EF10%值为16.6、9.3和5.5,分别比SPOT-Ligand 2和PoLi高出约10.7%、27.4%、17.0和9.2%、29.2%、17.0%.同时,在数据集DUD40上,Screener的EF值显著高于AutoDock Vina、FINDSITEcomb和SPOT-Ligand.在DUD-E65数据集上,Screener的EF1%值为25.7,EF5%值为10.9,EF10%值为6.1.尽管在DUD-E65上,Screener的EF1%稍低,但Screener的EF5%和EF10%值比SPOT-Ligand 2高出26.7%和19.6%.总体而言,本文所提出的Screener方法在数据集DUD-E65上明显优于AutoDock Vina,PoLi和SPOT-Ligand.例如,与SPOT-Ligand相比,Screener在EF1%、EF5%和EF10%上分别提高了91.8%、109.6%和48.8%.
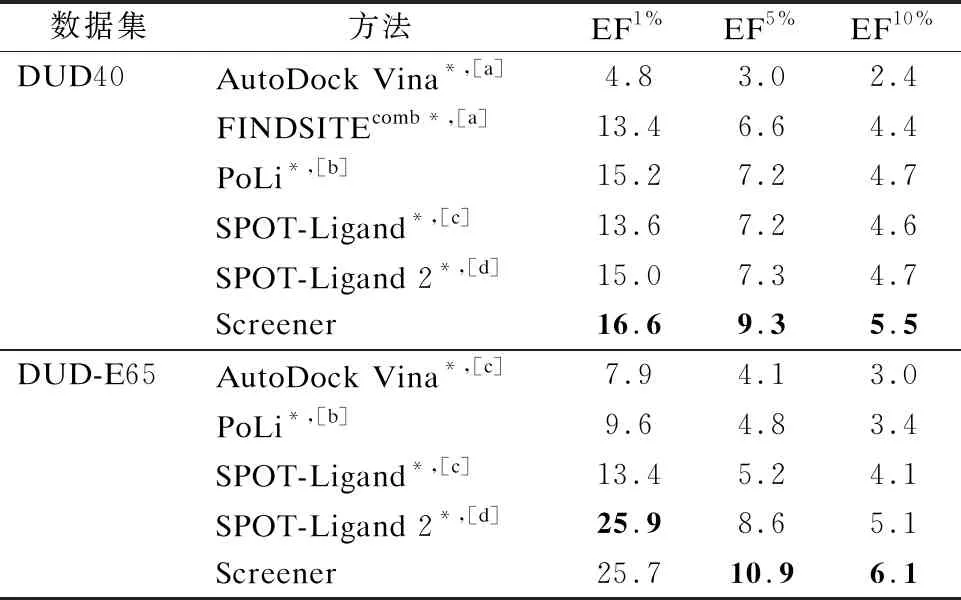
表3 Screener和基于真实结构的其他虚拟筛选方法在DUD40和DUD-E65数据集上的平均EF性能比较
结果节选自[a][13]、[b][14]、[c][15]、[d][10],*这些方法需蛋白质三维结构.
在PoLi中,活性分子结合口袋信息被认为可以改善筛选性能,然而,由于PoLi必须具有精确的受体蛋白质结构,所以对于没有受体蛋白结构的数据库,PoLi方法将无法使用,从而错过很多潜在的种子分子.在SPOT-Ligand 2中,为了使用没有受体蛋白质结构的数据库,它使用SPARKS-X[39]来预测受体蛋白质结构,然而,SPOT-Ligand 2的筛选性能很大程度上依赖于预测结构的质量,由于从头折叠模拟能力有限,预测结构的质量通常会随着目标蛋白值的序列长度增大而下降[40],更糟糕的是,受体蛋白质的活性分子结合位点信息被SPOT-Ligand 2忽略了.
总体而言,无论是使用受体蛋白质的建模结构还是生物实验测定的真实结构,相较于其他基于受体蛋白质结构的分子虚拟筛选方法,Screener实现了更好的筛选性能.主要原因在于:1)使用I-LBR对受体蛋白质进行特异性结合位点残基预测,这对于未知结合位点残基的蛋白质而言,I-LBR可以预测蛋白质的特异性潜在结合位点残基,从而提升分子筛选性能;2)通过蛋白质序列比对方法收集到了更多的种子分子,利用这些种子分子对待筛选分子集进行排序筛选,从而提升了分子虚拟筛选性能.
4 结 论
本文从受体蛋白质的序列出发,提出了一种名为Screener的基于蛋白质序列比对和活性分子相似性评估的分子虚拟筛选方法.对任一待筛选活性分子的受体蛋白质序列,Screener首先生成其PSFM特征、PSS特征以及PSA特征,并利用一种基于蛋白质序列的结合位点残基预测方法I-LBR[22]来预测受体蛋白质与分子的结合位点残基;其次,根据预测的结合位点残基以及其对应特征来搜索BioLiP[23]、DrugBank[24]和BindingDB[25]3个蛋白质与分子相互作用的数据库,构建待筛选蛋白质特异性的模板蛋白质库;然后,将所有与任意模板蛋白质相互作用的活性分子收集起来构建潜在的种子分子库;最后,利用分子2D指纹之间的相似性来对待筛选分子集进行排序,筛选出受体蛋白质的活性分子.在基准测试集DUD40和DUD-E65上的实验结果验证了Screener的有效性.为了进一步提高分子虚拟筛选的性能,在今后的研究中会着重研究两个方向:
1)开发更具有区分性的序列特征,改进蛋白质序列比对与匹配打分算法,更好地构建模板蛋白质库;
2)利用深度学习算法构建深度卷积神经网络,更精准地预测蛋白质的潜在结合位点残基.
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
中国听力语言康复科学杂志(2021年6期)2021-12-21
电脑报(2020年40期)2020-11-06
电脑知识与技术(2018年19期)2018-11-01
雪莲(2017年2期)2017-05-12
环球市场信息导报(2017年1期)2017-04-08
职业·下旬(2009年8期)2009-10-12
数理化学习·高一二版(2009年2期)2009-03-30
浙江中医杂志(2004年3期)2004-11-20
