
应变对(Ga,Mo)Sb 磁学和光学性质影响的理论研究*
2022-05-26 09:19潘凤春林雪玲王旭明
物理学报 2022年9期
潘凤春 林雪玲 王旭明
(宁夏大学物理与电子电气工程学院,银川 750021)
近年来,作为一种自旋电子学领域的关键材料,具有高温本征铁磁性的稀磁半导体受到了广泛的关注.为探索能够提高本征铁磁性居里温度(Curie temperature,TC)的方法,本文运用第一性原理LDA+U 方法研究了应变对Mo 掺杂GaSb 的电子结构、磁学及光学性质的影响.研究结果表明:–6%—2.5%应变范围下GaSb 半导体材料具有稳定的力学性能,压应变下GaSb 材料的可塑性、韧性增强,有利于GaSb 半导体材料力学性能的提升;应变对Mo 替代Ga 缺陷(MoGa)的电子结构有重要的影响,–3%至–1.2%应变范围下MoGa处于低自旋态(low spin state,LSS),具有1 µB 的局域磁矩,–1.1%—2%应变范围下MoGa 处于高自旋态(high spin state,HSS),具有3 µB 的磁矩;不管是LSS 还是HSS,MoGa 产生局域磁矩之间的耦合都是铁磁耦合,但铁磁耦合的强度和物理机制不同,适当的压应变可有效提高铁磁耦合强度,这有利于实现高TC 的GaSb 基磁性半导体;Mo 可极大提高GaSb 半导体材料的电极化能力,这有利于光生电子-空穴对的形成和分离,提高掺杂体系对长波光子的光电转化效率;Mo 引入的杂质能级使电子的带间跃迁对所需要吸收光子的能量变小,掺杂体系光学吸收谱的吸收边发生了红移,拉应变可进一步提升(Ga,Mo)Sb 体系在红外光区的光学性能.
1 引言
作为自旋电子学器件的关键材料,磁性半导体同时利用了电子的电荷属性和自旋属性而具有本征的半导体性质和铁磁性,已受到了广泛而持续的关注[1−7].其中III-V 族磁性半导体很容易与IIIV 族非磁性半导体GaAs,AlAs,GaP,InP 和InSb等结合形成异质结构,呈现出自旋相关的散射、层间相互耦合作用、隧穿磁阻等现象.有研究表明III-V 族GaSb 基磁性半导体具有可能高的TC、优异的电、磁、光等物理特性,从而成为磁性半导体中研究的热门材料[8−11].第一过渡族金属(V,Cr,Mn,Fe,Co,Ni 等)通常被选为III-V 族和氧化物等磁性半导体的掺杂剂[12−14],一方面是其3d 电子壳层是部分占据的,可提供局域磁矩,另一方面这些过渡族金属元素的原子半径和金属阳离子的半径相匹配,很容易形成替代掺杂.Dietl 等[2]利用平均场理论预测当Mn 掺杂含量和空穴浓度达到一定水平时,GaAs 基磁性半导体的TC可以提高到室温以上.Sato 等[15]从理论上预测Mn 掺杂GaSb 基磁性半导体的TC与Mn 的掺杂浓度成正比.Tu 等[16−18]在实验上证实Fe 掺杂GaSb 基磁性半导体的TC随着Fe 掺杂浓度的增大而升高,当Fe 掺杂浓度达到25%时,(Ga,Fe)Sb 的TC达到了340 K.由此可见,增大掺杂浓度是提高GaSb基磁性半导体TC的有效手段,为了进一步提高过渡族金属的掺杂浓度,在样品的制备过程中需要利用高温高压等极端条件对样品的生长氛围进行控制[19,20].然而,过高的掺杂浓度容易在样品中形成过渡族金属团簇及过渡族金属化合物等磁性第二相[21,22],因此有必要寻找另外的方法实现具有高TC和本征磁性的GaSb 基磁性半导体.从原子半径匹配的角度考虑,第二过渡族金属如Zr,Nb,Mo 等,更容易替代半导体中的金属阳离子形成替代掺杂,并且这些过渡族金属相是非磁性的,排除了磁性半导体中磁性来源于金属团簇的可能性.Medvedeva[23]利用第一性原理计算的方法,研究了Mo 掺杂In2O3的电子结构和磁光特性,揭示了磁相互作用在导电性以及控制光吸收的Burstein-Moss 位移方面的重要作用.Park 等[24,25]在Mo 掺杂的In2O3半导体中观察到了室温铁磁性.Egbo等[26]研究发现Mo 掺杂的In2O3体系位于导带底部电子的有效质量变小,这提高了载流子的迁移率.Lu 等[27]在空气中烧结的LaMn0.96Mo0.04O3中观察到的TC为238 K,认为样品的铁磁性来源于由Mo6+替代Mn3+后诱发的Mn3+和Mn2+之间的双交换作用.Dwivedi 等[28]第一次在Mo 掺杂的CoFe2O4中发现了磁有序和铁电性共存的现象,掺杂体系的磁化强度随着Mo 掺杂浓度的增大而增强,并把掺杂体系的巨介电常数归因于Maxwell-Wagner 弛豫机制.可以看出,Mo 作为一种掺杂元素可以实现半导体的铁磁性,并改善掺杂体系介电常数和光学吸收谱等光学性能.最近有研究表明[29−34],通过对半导体材料施加应变来调节其物理特性是一种可行的方法.Linpeng 等[29]指出应变对受主束缚空穴自旋弛豫和退相具有重要的作用.在双层膜中,研究者们发现应变可以诱导材料从半导体态到金属态的转变[31].(Ga0.8,Fe0.2)Sb 材料中的磁晶各向异性表现出了对外延应变的依赖性,并且当应变由拉应变变为压应变时其正负性发生了改变[32].通过选择不同衬底生长的半导体异质结构,在其内部往往具有拉应变或压应变,研究应变对半导体物理特性的影响具有重要的意义.作为III-V 族半导体材料,GaSb 具有禁带宽度小、电子和空穴迁移率高等优点,使其在半导体自旋电子器件、热光伏电池、红外激光器、红外探测器等领域有重要的应用前景.在GaSb 基磁性半导体领域,存在掺杂元素种类少(主要为Mn,Fe 掺杂),磁矩起源及磁相互作用机制并没有得到充分研究,研究结论比较浅显等显著问题.基于以上事实,本文研究了应变对Mo 掺杂GaSb 半导体的电子结构、磁学性质及光学性质的影响,探讨应变及Mo 掺杂对GaSb 半导体物理特性影响的微观物理机制,为实验上制备集磁、光功能于一体的新型半导体器件提供一定参考.
2 研究方法与模型的构建
本文采用基于密度泛函理论的CASTEP[35−37]软件进行研究.计算体系为64 个原子GaSb 超晶胞,包含32 个Ga 原子和32 个Sb 原子.电子体系波函数采用平面波波函数展开,平面波波函数截止能量为380 eV,计算体系电荷密度和总能量在布里渊区积分进行,基态能量采用Pulay 密度混合法,自洽精度为 5.0×10−7eV/atom,交换关联泛函采用LDA-CA-PZ 泛函,K空间网格点采用Monkhorst-Pack[38,39]方案,数值取为 3×3×3 .采用LDA+U方案来修正LDA/GGA 交换关联泛函对GaSb 半导体禁带宽度低估的这一问题,对Ga-3d 电子和Sb-5p 电子的库仑位能进行修正,当库仑位能的数值为UGa-3d=2.5 eV 和USb-5p=3.0 eV时计算得到的结果同实验值符合最好,此时得到的晶格常数a0=5.912 Å (实验值为6.096 Å,1 Å=0.1 nm),禁带宽度Eg=0.809 eV (实验值为0.810 eV)[40−42].
Mo 掺杂GaSb 有两种基本的缺陷类型,分别是MoGa和Mo 替代Sb 缺陷(MoSb).通常用缺陷的形成能来衡量晶体中缺陷的浓度含量,文中两种替代缺陷的形成能可由下式计算[43]:
Eformation=Edefect−Eperfect−µMo+µX,
式中Edefect表示包含一个MoGa(MoSb)的超晶胞的总能量,Eperfect表示完好的GaSb 超晶胞的总能量,µMo表示Mo 原子的化学势,µX表示Ga 原子或Sb 原子的化学势.缺陷浓度C与形成能Eformation的关系表达式为[43]
C=NsitesNconfigexp[−Eformation/(kBT)],
式中Nsites为单位体积晶格中缺陷位置的数目,Nconfig为产生缺陷的等效数量,kB是玻尔兹曼常数,T是热力学温度.由上式可知,当T一定时,缺陷浓度与形成能的大小有关,某一缺陷的形成能越小,相应的缺陷浓度就越大.计算结果表明,MoGa的形成能是1.576 eV,MoSb的形成能是3.606 eV,显然在热平衡状态下,MoGa的浓度高于MoSb.实验上和理论上已经证实[17,18,44,45],MoGa为主的掺杂GaSb 体系可以稳定存在,因此本文主要对MoGa的电子结构、磁学和光学特性进行理论计算研究.图1 给出了包含64 个原子的GaSb 超晶胞结构示意图,图中阿拉伯数字表示MoGa的位置,包含MoGa的GaSb 体系记为(Ga,Mo)Sb.本文应变的定义为

图1 GaSb 超晶胞结构,其中大的绿色球代表Ga 原子,小的紫色球代表Sb 原子Fig.1.Structure of GaSb supercell,where the big green balls and small purple balls denote Ga and Sb atoms,respectively.

式中a和a0分别表示有应变时和无应变时的晶格常数,ε>0 表示拉应变,ε<0 表示压应变.
3 计算结果和分析
3.1 应变下GaSb 的力学特性
材料结构的稳定性、刚性、韧性等力学特性取决于材料对外力的响应,因此研究材料的弹性性质是研究材料本构关系的重要内容.材料的体积模量(bulk modulus,B)和剪切模量(shear modulus,G)通常有两种不同的计算方法,分别是Voigt 提出的晶体边界上的应力连续模型[46]和Reuss 提出的晶体边界上的应变连续模型[47,48].Hill[49]证明了Voigt 模型和Reuss 模型的计算结果分别对应晶体弹性系数的上下限,并提出了晶体弹性系数取Voigt 模型和Reuss 模型计算结果算术平均值的Voigt-Reuss-Hill 模型.在弹性变化范围内,晶体的应力和应变成正比,弹性模量矩阵是对称矩阵,满足Cij=Cji.GaSb 半导体属于对称性最高的立方晶系,独立变化的张量元素个数为3 个,分别为C11,C44和C12.Voigt-Reuss-Hill 模型下体积模量和剪切模量的计算公式及力学稳定性判据(criteria,CRIT)如下:

图2 给出了–14%—5%应变区间力学稳定性判据的变化趋势,可以看出当应变范围为–6%—2.5%时,GaSb 半导体具有稳定的力学性能.表1列出了–6%—2%应变范围下GaSb 半导体的体积模量、剪切模量、泊松比和B/G的变化数据.
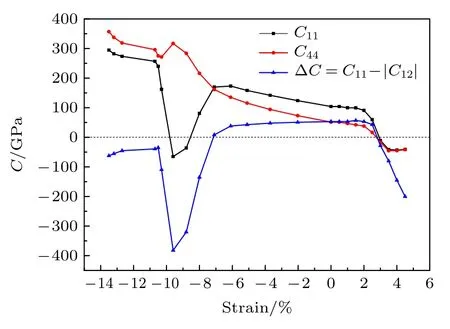
图2 力学稳定性判据Fig.2.Criteria of mechanical stability.

表1 应变下GaSb 的体积模量B、剪切模量G、泊松比 γ 和B/GTable 1.Bulk modulus B,shear modulus G,Poisson ratio γ and B/G under strains.
体积模量反映了材料在弹性系下对外界均一性压缩的抵抗能力.由表1 可以看出,GaSb 材料的体积模量随着压应变的增大而增大,随着拉应变的增大而减小.晶体的体积模量与组成晶体化学键的伸缩能力有关,微观水平下晶体的体积模量由化学键的强度和压缩性来决定[50].计算结果表明,随着压应变的增大,Ga 原子的失电子能力逐渐减弱,Ga—Sb 化学键的键布居逐渐增大,这说明Ga—Sb化学键的离子性减弱,共价性增强.从另一个角度看,原子抓电子能力越强,价电子云在外力作用下越难发生移动,因此一定程度上可以认为化学键的共价性越强,材料对外界压缩的抵抗力也就越强.同时从表1 可以看出,剪切模量在零应变状态下的数值最大为47.21,且随着拉应变或压应变的增大而变小,表示材料在应变下抗剪切形变的能力逐渐减弱,可塑性增强.
泊松比表示纵向应力所引起的横向应变与相应纵向应变之比的绝对值,是反映材料横向变形的弹性常数.从表1 可以看出,GaSb 材料的泊松比γ数值随着压应变的增大而逐渐增大.按照断裂行为的判据[51],泊松比高的材料具有高的韧性,因此可认为随着压应变的增大,GaSb 半导体材料的韧性增强,发生脆性断裂的可能性小.根据Pugh 判据[52],B/G可以表示材料的塑性变形能力,从表1可以看出,B/G随着压应变或拉应变的增大而逐步升高,说明应变下GaSb 半导体材料的延展性变好.
综上所述,–6%—2.5%应变范围内,GaSb 半导体材料具有稳定的力学性能.在此应变范围内,随着压应变的增大,GaSb 材料的抗压性增强,抵御塑性形变的能力降低,材料具有较好的可塑性.泊松比的数值表明,压应变下GaSb 半导体材料的韧性增强,发生脆性断裂的可能性较小.可以看出,适当的压应变有利于GaSb 半导体材料力学性能的提升,这对GaSb 半导体在极端环境下保持物理性能的稳定性是有利的.
半导体材料在制备的过程中,一般可通过材料设计,包括引入缺陷[53,54],构建失配的异质结构[55,56]等方法在材料的内部引入压应变或拉应变.王志伟等[57]利用等离子体增强化学气相沉积法在晶格失配较大的GaSb 衬底上沉积SiO2薄膜,通过改变薄膜沉积时的工艺条件,如反应温度、射频功率、反应压强等获得了具有不同内部应变的SiO2薄膜.Goel 等[32]利用分子束外延法在不同缓冲层上制备了具有不同应变的(Ga,Fe)Sb 磁性半导体材料(ε=−1.7 %,缓冲层AlSb;ε=0.23 %,缓冲层In0.5Ga0.5As;ε=3.84 %,缓冲层GaAs),并认为通过不同的缓冲层可生长出具有不同应变的(Ga,Fe)Sb 磁性半导体材料.Kondrin 等[19]在实验上研究了(Ga,Cr)Sb 体系在高压(6—8 GPa)下的电子输运特性和磁特性,第一性原理计算结果表明,当施加静水压力为8 GPa 时,GaSb 体系的应变可以达到ε=−3.5 %,可见对样品施加高压也是使其内部产生压应变的有效手段.基于半导体材料晶格常数的考虑,可认为通过以AlSb,CdSe,InSb等(InAs,InP,AlAs 等)作为缓冲层可制备出具有不同拉应变(压应变)的(Ga,Mo)Sb 半导体材料.考虑到太大的拉应变或压应变在材料制备的过程难以实现,本文主要研究–3%—2%应变范围内(Ga,Mo)Sb 体系的磁学及光学性能.
3.2 应变下(Ga,Mo)Sb 的磁学特性
研究了不同应变下包含一个MoGa的(Ga,Mo)Sb 体系的电子结构和磁特性.计算结果表明,当应变在–1.1%—2%范围内变化时,(Ga,Mo)Sb体系的态密度图(density of states,DOS)和磁矩的分布具有相似性.图3(a)和图3(b)给出了零应变下(Ga,Mo)Sb 体系的总态密度图(total DOS,TDOS)和Mo-4d 电子的投影态密度图(projected DOS,PDOS).从图3(a)可以看出,零应变下(Ga,Mo)Sb 体系的自旋向上电子和自旋向下电子的态密度在零点费米能级处不对称,发生了自旋劈裂,说明(Ga,Mo)Sb 体系中产生了净磁矩.图3(b)给出了Mo-4d 电子的PDOS,Mo-4d 电子占据自旋向上态的电子数目多于占据自旋向下态的电子数目,表明(Ga,Mo)Sb 体系中的Mo 原子产生了自旋向上(正)的净磁矩.图4(a)给出了不同应变下(Ga,Mo)Sb 体系中磁矩的分布来源,计算结果显示,零应变下Mo 原子产生3.04µB的磁矩,近邻Sb 原子产生–0.13µB的磁矩,其余磁矩由次近邻Ga 原子所贡献,一个MoGa产生总磁矩的大小为3µB,可以看出磁矩主要由掺杂Mo 原子所贡献.为了形象展示磁矩的这一分布,图5(a)给出了–1%应变下包含一个MoGa的(Ga,Mo)Sb 体系的空间自旋密度分布图,图中的蓝色部分表示由Mo 原子产生向上的净自旋,黄色部分是Mo 近邻4 个Sb 原子产生向下的净自旋.–1.1%—2%应变范围内,一个MoGa产生总的磁矩都为3µB,只是磁矩的分布有所不同,见图4(a),此时MoGa处于高自旋态(high spin state,HSS).
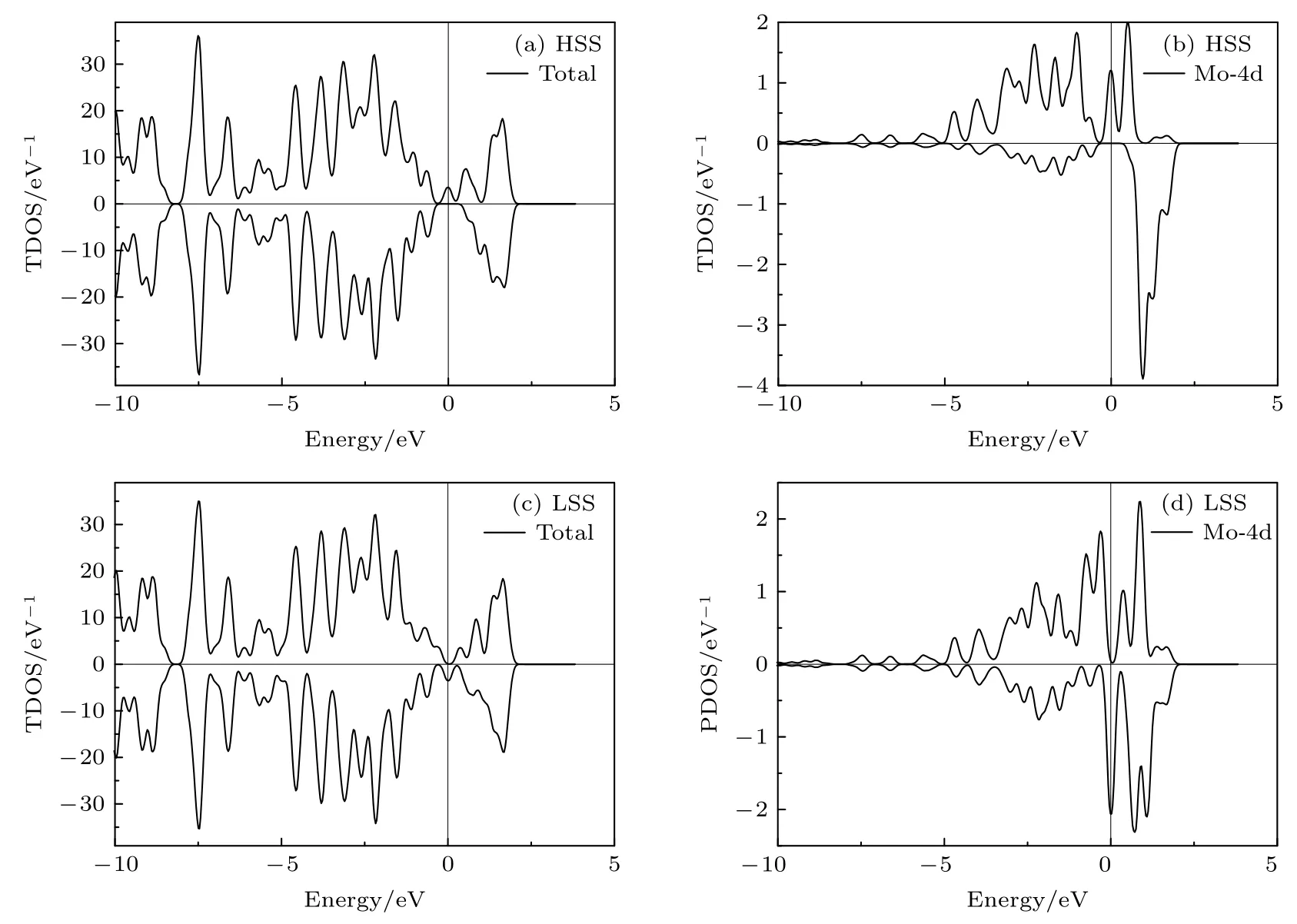
图3 (Ga,Mo)Sb 体系在HSS 和LSS 下的态密度图 (a) 零应变下的TDOS;(b)零应变下Mo-4d 电子的PDOS;(c)–1.2%应变下的TDOS;(d)–1.2%应变下Mo-4d 电子的PDOSFig.3.Density of states (DOS) of (Ga,Mo)Sb in HSS and LSS:(a) TDOS and (b) PDOS of Mo-4d without strain;(c) the TDOS and (d) the PDOS of Mo-4d under–1.2% strain.
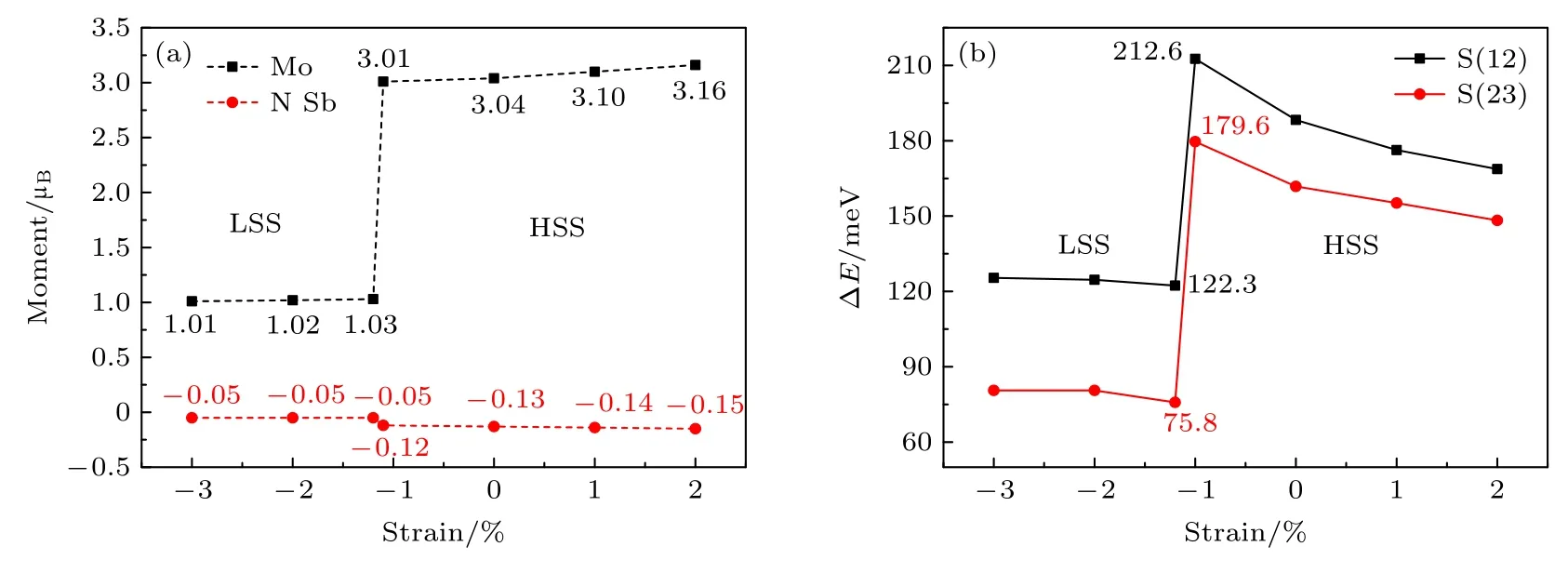
图4 (a) 应变下,(Ga,Mo)Sb 体系中掺杂Mo 原子和近邻Sb 原子的磁矩;(b) 应变下,S(12)和S(23)结构的ΔEFig.4.(a) Magnetic moments contributed by Mo and neighbor Sb (N Sb) under strains and (b) ΔE of S(12) and S(23) under strains,respectively.
当应变在–3%至–1.2%范围变化时,MoGa转变为低自旋态(low spin state,LSS),此时(Ga,Mo)Sb 体系产生的总磁矩为1µB.从图3(c)可以看出(Ga,Mo)Sb 体系的TDOS 自旋向上电子占据数目和自旋向下电子占据数目不相等,表明体系中产生了净磁矩.图3(d)给出了Mo-4d 电子的PDOS,同样表明掺杂体系中Mo 原子产生了正的净磁矩.比较图3(b)和图3(d)可以看出,HSS 下Mo-4d 电子在零点费米能级处的一个自旋向上的态密度峰是部分占据的,而LSS 下Mo-4d 电子在零点费米能级处有一个电子部分占据的自旋向下的态密度峰.从图4(a)可以看出,–3%至–1.2%应变范围下(Ga,Mo)Sb 体系的磁矩主要由Mo 原子所贡献,Mo 原子近邻Sb 原子产生自旋向下的净磁矩,且数值很小.LSS 下MoGa的空间自旋密度分布图,见图5(b),可以看出,Mo 原子周围4 个近邻Sb 原子产生的磁矩很小.

图5 等值面为0.01 e/Å3 的空间自旋密度图 (a) HSS下MoGa;(b) LSS 下MoGa;(c) HSS 下S(12)结构FM 耦合;(d) LSS 下S(23)结构FM 耦合Fig.5.The spatial spin density for (a) MoGa under HSS,(b) MoGa under LSS,(c) FM coupling of S(12) under HSS,(d) FM coupling of S(23) under LSS,respectively.The isovalue is set to 0.01 e/Å3.
显然,掺杂的Mo 原子在一定程度上诱导了GaSb 半导体的自旋极化,应变对MoGa产生的局域磁矩大小和分布都有影响.MoGa中,Mo 原子周围最近邻的4 个Sb 原子构成正四面体,Mo 原子位于正四面体的中心.以正立方体为参照系,坐标原点位于正立方体的中心,坐标沿着与正立方体的棱平行的方向伸展,见图6.4 个最近邻Sb 原子位于正立方体互不相邻的4 个顶点上,dxy,dxz和dyz这3 条d 电子轨道的波瓣指向正立方体各棱的中心,距离4 个Sb 原子较近,受到较大的排斥作用使轨道能量升高较多,分裂后这3 条d 电子轨道的能量简并,用群论的符号记为 t2轨道.另外两个轨道的波瓣指向正立方体的面心,距离4 个Sb 原子较远,受到的排斥力较小,轨道能量升高较少,分裂后这两条轨道的能量简并,记为e 轨道.四面体晶场作用下d 电子轨道的分裂能即为 t2轨道和e 轨道之间的能量差,记为Δt.
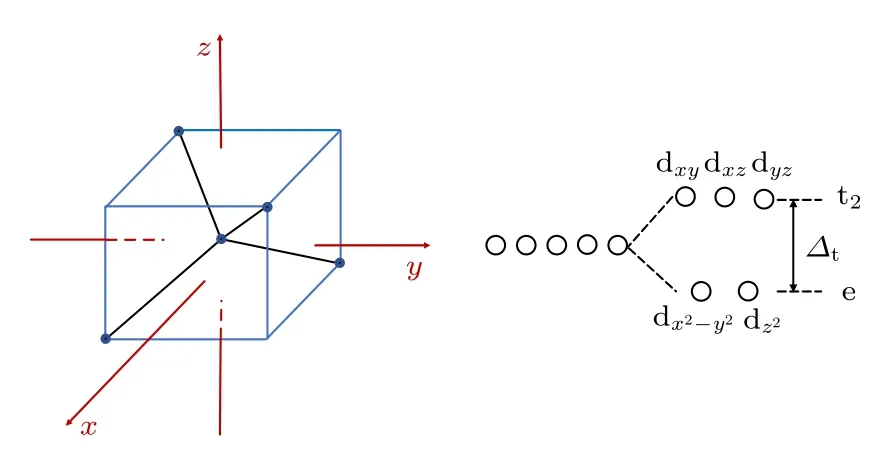
图6 d 电子轨道四面体晶场分裂示意图Fig.6.Diagram of Mo-4d orbital splitting under tetrahedral crystal field.
Mo 原子的电子组态是[Kr]4d55s1,当Mo 原子替代Ga 原子后,Mo 原子失去3 个价电子与周围近邻的4 个Sb 原子成键,此时Mo 离子的电子组态变为[Kr]4d3.Mo-4d 电子轨道在四面体晶场的作用下分裂成能量较低的e 轨道和能量较高的t2轨道,Mo 剩余的3 个4d 电子的排布有两种可能:2 个电子排在自旋向上的e 轨道,1 个电子排在自旋向下的e 轨道,电子结构可以表示为 e↑↑↓(↑表示自旋向上的电子,↓表示自旋向下的电子),这种方式称为LSS,此时电子成对要克服成对能EP;为了避免成对能,2 个电子在自旋向上的e 轨道,1 个电子在自旋向上的 t2轨道,电子结构可以表示为,这种方式称为HSS.显然,当Δt>EP时电子排布采取LSS,反之采取HSS.
–1.1%—2%应变范围下,Mo-4d 电子的轨道分裂能Δt

图7 (Ga,Mo)Sb 体系的能带图 (a)零 应 变 下HSS;(b)–1.2%应变下LSS.黑色实线表示自旋向上的能带,红色虚线表示自旋向下的能带.在能量为0 处的水平蓝色虚线表示费米能级Fig.7.Band structures of (Ga,Mo)Sb in (a) HSS without strain and (b) LSS under–1.2% strain.Black solid lines and red dotted lines denote spin-up and spin-down bands,respectively.The horizontal blue dotted lines located at 0 energy are Fermi levels.
为了研究MoGa产生局域磁矩之间的耦合特性,在包含64 原子GaSb 超晶胞结构中引入两个MoGa,为了避免第一性原理计算中镜像缺陷之间不合理的相互作用对计算结果的影响,两个MoGa之间的距离不应该超过掺杂体系晶格常数的一半.设计了两种结构:用两个Mo 原子替代图1 中的1 号和2 号Ga 原子,记为结构S(12);用两个Mo 原子替代2 号和3 号Ga 原子形成结构S(23).零应变下,S(12)和S(23)结构中两个Mo 原子之间的距离分别为4.180 Å和5.912 Å,其中S(23)结构中两个MoGa都在(010)晶面内.设定两种结构下Mo 原子的初始自旋,就可以计算铁磁(ferromagnetism,FM)耦合和反铁磁(anti ferromagnetism,AFM)耦合状态下体系的总能量,MoGa的磁耦合特性可以根据FM 状态下体系总能量(EFM)和AFM 状态下体系总能量(EAFM)的能量差来判断,ΔE=EAFM−EFM,若 ΔE >0,表明MoGa产生的局域磁矩倾向于FM 耦合,反之为AFM 耦合.
图4(b)给出了 ΔE随应变的变化趋势,ΔE可以表征局域磁矩之间的耦合强度和相应磁序的稳定性.从图4(b)可以看出,S(12)和S(23)两种结构在不同应变下,ΔE >0,ΔE越大表明体系FM 耦合相互作用越强,TC越高[58].图4(b)的结果表明,无论是LSS 还是HSS,掺杂体系局域磁矩之间的耦合都倾向于FM 耦合,其FM 耦合机制可以用d-d 交换模型和p-d 交换模型来解释[59].–3%至–1.2%应变范围下MoGa处于LSS,其电子结构为 e↑↑↓,e−轨道跨过零点费米能级,见图3(d)或图7(b),不同MoGa的 e−轨道上电子之间的交换并不需要改变电子的自旋方向,因此LSS 下MoGa产生局域磁矩之间的耦合是FM 耦合,这种由Mo-4d 电子直接交换产生的相互作用称为d-d 交换作用.–1.1%—2%应变范围下MoGa处于HSS,其电子结构为,t2+轨道跨过零点费米能级,见图3(b)或图7(a),t2+轨道上电子之间的交换作用(d-d 交换作用)导致MoGa产生的局域磁矩之间的耦合同样是FM 耦合,虽然HSS 下e 轨道是半占据的,但由于 e+轨道位于零点费米能级下–1.25 eV处,见图7(a),因此 e+轨道上的电子并不会产生直接的交换作用.
图4(b)中的两条 ΔE曲线展现了左低相差较大和右高相差较小的特点.从图4(b)可以看出,–3%至–1.2%应变范围下,S(12)结构的 ΔE(黑色方块线)较小,–1.2%应变下的数值为122.3 meV,当应变增大到–1%时,ΔE跃变到212.6 meV.对比图4(a)可以看出,MoGa也由LSS 转变到HSS.对于S(23)结构,–3%至–1.2%应变范围内的ΔE(红色圆点线)低于同应变范围内S(12)结构的 ΔE,–1.2%应变下的数值只有75.8 meV,当应变增大到–1%时,ΔE跃变到179.6 meV,此时S(23)结构中MoGa由LSS 转变到HSS.S(12)和S(23)两种结构在LSS 下(–3%至–1.2%)的 ΔE均明显小于其在HSS 下(–1%—2%)的 ΔE,这是因为LSS下局域磁矩之间相互作用只能通过d-d 交换作用来实现,但对于HSS 状态,除了d-d 交换作用,还有Mo-4d 电子通过近邻Sb-5p 电子产生的间接交换作用,这种交换作用称为p-d 交换作用.图4(a)的计算结果表明,LSS 下掺杂Mo 原子近邻Sbp 电子产生的磁矩(–0.05µB)远小于HSS 下近邻Sb-p 电子产生的磁矩(–0.12µB),因此HSS 下局域磁矩之间的FM 耦合是d-d 交换作用和p-d 交换作用共同支配的结果,HSS 下FM 耦合相互作用更强一些,这导致了较大的 ΔE.对于LSS,相同应变下S(12)和S(23)两种结构体系的 ΔE相差较大,这是由d-d 交换作用的短程性的特点所导致[4],S(23)结构下两个MoGa的距离较远,d-d 交换作用的强度迅速减弱.对于HSS,相同应变下S(12)和S(23)两种结构体系的 ΔE变化相对较小,这是因为HSS 下除了d-d 交换作用,还有p-d 交换作用,而p-d 交换作用具有长程性[4],因此HSS 下MoGa的间距对其产生局域磁矩之间耦合强度的影响没有LSS 下那样明显.值得关注的是,在–1.1%—0%应变范围内,Mo 处于HSS,此时局域磁矩之间的FM 耦合强度明显大于零应变时的情况,说明在此应变范围内对实现高TC本征铁磁性(Ga,Mo)Sb半导体是极其有利的.
对于III-V 族磁性半导体各种磁现象的解释,Dietl 等[2,60]建立了空穴(hole,h)载流子导致局域化自旋磁性相互作用的平均场模型.从图4(a)可以看出,–1.2%应变下Mo 产生的磁矩为1.03µB,–1%应变下Mo 产生的磁矩为3.01µB,因此(Ga,Mo)Sb 体系中Mo 原子价态为+3 价,此时Mo-4d 电子数目为3 个.根据Hund 定则,半满的d 壳层(5 个d 电子)是比较稳定的,因此MoGa中心将束缚2 个电子在4d 壳层,再加上2 个弱束缚的空穴构成的状态比较稳定,此时MoGa的电子态为A0(d5+2h),这里A0表示电中性中心,这样Mo 原子替代3 价Ga 原子,既提供了局域化的自旋,又提供了空穴载流子,由于p-d 杂化,Mo 杂质带附近的价带空穴自旋与Mo 局域自旋方向相反.由于价带波函数比较扩展,一个空穴可以与一定数目的Mo 局域自旋结合,当空穴浓度足够高时,就可以产生以空穴载流子为媒介的自旋与自旋之间的相互作用.在(Ga,Mn)As 磁性半导体中,Mn的价态为+3 价,Mn 替代Ga 缺陷(MnGa)中心的电子态为A0(d5+h)[61,62],可以看出,相比于Mn 原子,Mo 原子在增大空穴载流子浓度方面更具有优势,这对于提高GaSb 基磁性半导体的TC是极为有利的.
图5(c)给出了HSS 下S(12)结构两个MoGa产生局域磁矩FM 耦合的自旋密度空间分布图.可以看出磁矩之间的相互作用是通过Mo-4d 电子波函数的交叠(d-d 交换作用)和Sb-5p 电子波函数的交叠(p-d 交换作用)来实现.比较图5(a)和图5(c)可知,图5(c)中的两个MoGa均处于HSS.图5(d)给出了LSS 下S(23)结构两个MoGa产生局域磁矩FM 耦合的自旋密度空间分布图,LSS 下Sb-5p 电子对磁矩的贡献很小,MoGa产生磁矩之间的相互作用主要通过d-d 交换作用而非p-d 交换作用来实现.比较图5(b)和图5(d)可知,图5(d)中两个MoGa均处于LSS.
3.3 应变下(Ga,Mo)Sb 的光学特性
GaSb 由于其较低的禁带宽度,使其可以与多种辐射体的光谱相匹配,因此以GaSb 材料为基础的热光伏电池得到了广泛的研究[63,64].为了使GaSb基热光伏电池更好地与温度较低的辐射器相匹配,基于GaSb 相关的三元、四元III-V 族化合物被发现并用来制作热光伏电池.虽然多元GaSb 相关化合物的禁带宽度最低达到了0.53 eV,有效拓展了红外光谱的响应范围,但由于较大的表面复合速度而限制了热电转换效率的进一步提升[65−67].目前用于热光伏电池的GaSb 基半导体材料仍然存在禁带宽度偏大,不能很好地和热辐射器相匹配以及实际转换效率不高等问题,因此有必要研究(Ga,Mo)Sb 体系的光学性质,拓展GaSb 基半导体材料在红外热光伏电池、红外半导体激光器、探测器等领域的应用.
半导体在线性响应范围的光学性质通常用复介电函数ε(ω)=ε1(ω)+iε2(ω)来描述,其中ε2(ω)表示复介电函数的虚部,由价电子在占据轨道和非占据轨道之间的跃迁来计算;ε1(ω)表示复介电函数的实部,由ε2(ω)所满足的Kramers-Krönig 色散关系确定[68].具体函数表达式为
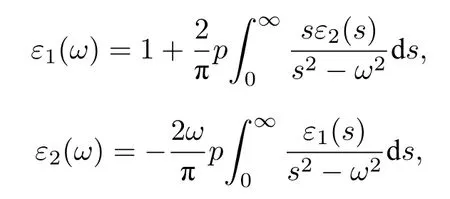
式中p为积分主值,ω为入射光子的圆频率,s为积分变量.积分主值p的表达式为
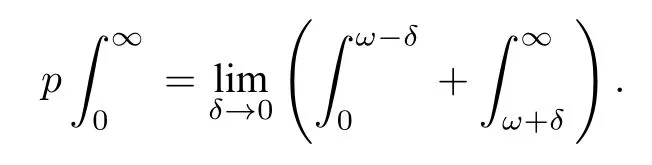
利用复介电函数与复折射率之间的函数关系((n(ω)+ik(ω))2=ε1(ω)+iε2(ω))和Kramers-Krönig 色散关系可以得到吸收系数α(ω)[68],函数表达式如下:
α(ω)=2ωk(ω)/c
式中c为光速,普通折射率n(ω)为复折射率的实部,决定光衰减的k(ω)为复折射率的虚部.
图8(a)给出了应变下GaSb 和(Ga,Mo)Sb的光学吸收谱.可以看出GaSb 体系(黑色实线)对远红外光区光子没有吸收,其光学吸收谱的吸收边落在了1667 nm 处,此波长光子对应的能量是电子从价带顶跃迁到导带底所需要的最小能量,即0.809 eV.–2%—2%应变范围内,(Ga,Mo)Sb体系在大于300 nm 范围内对入射光子的吸收幅度相比于未掺杂GaSb 体系均有很大的提升,且其光学吸收谱的吸收边落在了远红外光区,即发生了红移现象.从图7 可以看出,Mo 掺杂引入的杂质能级位于价带和导带之间,且跨过零点费米能级,电子由价带向导带跃迁时以杂质能级为桥梁,跃迁所需要的能量变小,因此提升了(Ga,Mo)Sb 体系对长波光子的吸收能力,这是(Ga,Mo)Sb 体系光学吸收谱的吸收边发生红移的原因.从图8(a)还可以看出,(Ga,Mo)Sb 体系对可见光区和红外光区光子的吸收幅度随着拉应变的增大而增大,随着压应变的增大而减小.这说明应变对(Ga,Mo)Sb体系的光吸收也有影响,适当的拉应变有利于提升(Ga,Mo)Sb 体系对可见光区和红外光区光子的吸收幅度.
复介电函数的实部ε1(ω) 表征了半导体材料在外电场作用下的极化程度,实部数值越大说明体系的电极化能力越强,在入射光子能量为零时对应的数值为静态介电常数.从图8(b)可以看出,零应变下GaSb 复介电函数实部(黑色实线)在入射光子能量为0 时的数值为19.43,零应变下(Ga,Mo)Sb体系(绿色实线)所对应的数值为32.84.这说明MoGa可以有效提高(Ga,Mo)Sb 体系的介电性能,掺杂体系电极化能力的提升和光生电场强度的变大有利于光生电子-空穴对的形成和分离,提高了掺杂体系对长波光子的光电转换能力.–2%—2%应变范围内,(Ga,Mo)Sb 体系在0—1.75 eV 区间的实部数值均大于零应变GaSb 体系所对应实部数值,且(Ga,Mo)Sb 体系的静态介电常数随着拉应变的增大而增大,随着压应变的增大而减小.这同样说明应变对(Ga,Mo)Sb 体系的光电转换效率有重要影响,适当的拉应变有利于(Ga,Mo)Sb 体系光学性能的提升.
复介电函数的虚部ε2(ω) 表征的是半导体内部形成电偶极子所消耗的能量,虚部数值越大说明价电子处于激发态的数目越多,反映了半导体材料中电子的受激跃迁程度.从图8(b)可以看出,–2%—2%应变范围内,(Ga,Mo)Sb 体系在0—1.51 eV能量区间的虚部数值均大于零应变下GaSb 体系(黑色虚线)对应能量区间的虚部数值,表明(Ga,Mo)Sb 体系对红外、远红外光区光子的吸收能力增强.零应变(Ga,Mo)Sb 体系的虚部(绿色虚线)在光子能量为0 的数值为0.87,而GaSb 体系对应虚部的数值为0.这说明(Ga,Mo)Sb 体系对能量为“0”的光子就有响应,其原因在于Mo 引入的杂质能级是部分占据的且跨过零点费米能级,电子在简并的杂质能级之间跃迁并不需要吸收光子即可发生,此即电子的带内响应,虽然Mo 掺杂引入杂质能级上电子的带内响应对掺杂体系的光学性质有一定的影响,起主要作用的是电子在不同能级之间的直接跃迁[69].–2%—2%应变范围内,(Ga,Mo)Sb 体系在低能区的数值随着拉应变的增大而增大,随着压应变的增大而降低,说明拉应变可以提升(Ga,Mo)Sb 体系电子的受激跃迁程度,有利于材料光催化性能的提升.

图8 (a) 应变下,GaSb 和(Ga,Mo)Sb 的光学吸收谱;(b) 应变下,GaSb 和(Ga,Mo)Sb 的复介电函数,实线代表实部,虚线代表虚部Fig.8.(a) Absorption spectra and (b) complex dielectric function of GaSb and (Ga,Mo)Sb under strains.In panel(b) solid and dotted lines denote real and imaginary parts,respectively.
(Ga,Mo)Sb 体系光学性质部分的计算模型为包含一个MoGa的64 个原子GaSb 超晶胞结构体系,此时掺杂Mo 原子的摩尔浓度为3.125%.若考虑(Ga,Mo)Sb 体系磁性半导体具有较高的TC,Mo 原子的掺杂浓度不应低于6.25%,这也正是S(23)结构中Mo 原子的掺杂浓度.虽然掺杂浓度高,费米能级处杂质能级上电子的数目多,有利于(Ga,Mo)Sb 体系对红外光区、远红外光区光子的吸收;但也有其不利的一面,由掺杂元素引入杂质能级的附加势是作用距离仅为一两个原子的短程势[68],高浓度掺杂下密集的Mo 原子可以成为电子-空穴对的有效复合中心,从而使载流子的寿命大大降低,影响材料的光学性能.采用低温分子束外延生长方法,浓度高达25%的(Ga,Fe)Sb 样品实验上已制备成功[16,17],理论上关于铁磁性计算的掺杂浓度高达50%[11,70],若综合考虑(Ga,Mo)Sb体系的磁学和光学性质,可认为在均匀掺杂前提下,提高掺杂浓度既可以增大体系中的空穴载流子浓度,又可以避免电子-空穴对复合中心的形成[69,71].
以上分析结果表明,MoGa的掺杂有利于提高GaSb 半导体材料中载流子的受激跃迁程度,同时掺杂体系的电极化能力和光生电场强度的增强有利于光生电子-空穴对的产生和分离,改善了(Ga,Mo)Sb 体系的光催化特性,提升了(Ga,Mo)Sb 体系对可见光区、红外光区光子的吸收幅度,拉应变可进一步提高(Ga,Mo)Sb 体系的光学性能.
4 结论
运用第一性原理LDA+U的方法研究了不同应变下Mo 掺杂GaSb 的电子结构、磁学和光学性质.结论如下:1)–6%—2.5%应变范围内GaSb半导体材料具有稳定的力学性能.随着压应变的增大,GaSb 材料的抗压性和可塑性增强,材料发生脆性断裂的可能性较小,适当的压应变有利于GaSb 半导体材料力学性能的提高,这对GaSb 半导体材料在极端环境下保持稳定的物理性能是有利的.2) 应变对MoGa的电子结构有重要的影响.–3%至–1.2%应变范围下MoGa处于LSS 并产生1µB的局域磁矩,局域磁矩之间的耦合是FM 耦合,强度较弱,FM 耦合的微观物理机制可用dd 交换作用模型来解释.–1.1%—2%应变范围下MoGa处于HSS 并产生3µB的磁矩,可用d-d 交换作用模型和p-d 交换作用模型来解释局域磁矩之间的FM 耦合,HSS 下磁矩之间的FM 耦合具有很高的稳定性.HSS 下的FM 耦合强度远大于LSS 下的FM 耦合强度,对于HSS,ΔE随着压应变增大而变大,–1%应变下的 ΔE高达212.6 meV,表明对于(Ga,Mo)Sb 体系适当的压应变可以使MoGa处于HSS,且MoGa产生局域磁矩之间的FM 耦合强度也会明显增强.3) MoGa引入的杂质能级可以作为电子从价带向导带跃迁的桥梁,电子的带间跃迁对所需要吸收光子的能量变小,从而提升了(Ga,Mo)Sb 体系对长波光子的吸收能力,光学吸收谱的吸收边落在了远红外光区,有效拓展了(Ga,Mo)Sb 体系对光子的吸收范围;零应变下,MoGa使GaSb 半导体的静态介电常数由19.43 提升至32.84,表明MoGa的引入可以大大提高(Ga,Mo)Sb 体系的电极化能力,有利于(Ga,Mo)Sb 体系光生电子-空穴对的产生和分离;(Ga,Mo)Sb 体系复介电函数虚部数值的增大表明其价电子具有很高的受激跃迁程度.MoGa提高了(Ga,Mo)Sb体系对长波光子的吸收及其光电转换效率,提高了材料的光催化特性.此外,拉应变可进一步提升(Ga,Mo)Sb 在红外光区的光学性能.
通过对应变下(Ga,Mo)Sb 体系磁学和光学性质影响的研究分析,我们认为在样品的生长过程中,施加一定的应变可提升(Ga,Mo)Sb 体系的磁学及光学性能,期望我们的研究为实验上制备新型的GaSb 基半导体材料提供一定的理论参考.
猜你喜欢
物理学报(2022年11期)2022-06-18
当代党员(2022年9期)2022-05-20
物理学报(2022年7期)2022-04-15
当代陕西(2022年1期)2022-03-09
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
华人时刊(2021年23期)2021-03-08
中国舰船研究(2020年3期)2020-06-29
中学生数理化·高三版(2017年1期)2017-04-20
