
活性中间体2,12-二甲氧基-7-氧代-[5]-螺旋烯的合成
2022-05-26 04:23:06张小敏
绵阳师范学院学报 2022年5期
陈 英,张小敏
(绵阳师范学院化学与化学工程学院,四川绵阳 621000)
0 引言
虽然螺旋烯(呈现螺旋状非平面骨架共轭分子)早已为人所知[1],但直到近来因发现其独特的结构和电子特性而引起一些化学家很大的兴趣。这些兴趣主要集中在探索其作为材料和催化科学以及医药中间体等方面的潜在应用.合成螺旋烯的原料联萘酚是一种重要的手性化合物,在有机合成、农药、染料特别是医药领域都有着极其重要的作用,据文献报道螺旋烯分子是联萘酚的两个酚羟基环化成醚得到的非对映体,还可以在BBr3、BCl3作用下,开环分别得到光学纯R-联萘酚和S-联萘酚,这对于手性药物的研究更是一个重大的突破[2].
本文所讨论的7-氧代-[5]-螺旋烯是一类联萘酚醚化合物,属于联芳香类化合物,其在合成药物、高分子材料和液晶等领域都有重要价值[3].目前,合成联芳香类化合物的方法归纳起来主要有三种:一是经过Suzuki,Kumada,Stiie和Negish等偶联反应来合成,该类反应都是通过以Pd等过度金属为催化剂来实现[4].这是一种非常经典的合成芳香类化合物的方法,该方法研究时间很长并且条件摸索的非常透彻,收率可观并且易于操作,因此广泛应用于实验室和工业生产当中.但该类方法有一个缺陷,不同的芳香化合物,性质存在差异,有些芳香化合物需要合成为相应的芳基硼酸、芳基硼酸酯或者卤化物等,这不仅延长了反应步骤,无形中增加了合成难度,还会造成溶剂以及相关原材料的过度消耗.二是采用卤化物、芳香硼等非官能团芳香化试剂通过偶联反应来制备联芳香化合物;三是在氧化剂作用下,将两个未官能团化芳香化合物的C-H键采用过渡金属活化,然后直接氧化偶联来制备联芳香类化合物,这也是最近发展起来的研究热点之一.Toda课题组报道以铁(Ⅲ)作为催化剂[5];后又有报道以FeCl3为催化剂,水为溶剂,利用微波辐射技术快速合成联萘酚,产率可达91%,而且反应时间短、原料不需要预处理、成本低,但是工艺不易放大[6].李德昌等利用萘酚和联萘酚在醇中溶解度的差异(萘酚易容于醇、联萘酚微溶),引入醇作为反应溶剂,并得到可行的工艺路径,与传统的液相法相比,具有原材料不需要精制、反应时间短、反应条件温和、收益良好、成本低等优点[7];后杨思君提出固相合成工艺,即将2-萘酚与铁(Ⅲ)催化剂混合均匀并充分研磨后放入烘箱进行反应,经后处理以及纯化后可以得到含量(HPLC)大于99%[8].除上述以铜盐(Ⅱ)、铁盐(Ⅲ)来合成外消旋联萘酚外,还有电化学法[9]、氧钒配合物为催化剂、溴为催化剂[10]、固体路易斯酸为催化剂[11]、FeCl3-CuCl2为催化剂[12]等方法.
本文主要对以2,7-二羟基萘为原料,首先通过克级小试不断摸索反应条件和纯化方法,最终完成百克级目标产物的合成并优化工艺对目标化合物的结构进行确定.
1 主要仪器、试剂
1.1 主要仪器
循环水多用真空泵(SHK-Ⅲ郑州科泰实验设备有限公司)、旋转蒸发仪(BC-R206上海贝凯生物化工设备有限公司)、暗箱式紫外分析仪(ZF-2D上海强运科技有限公司)、快速制备色谱仪(COMBIFLASH Rf 150PSI环球(香港)科技有限公司)、NMR Magnet System(400 MHz/54 mm布鲁克科技有限公司)、液相质谱联用仪(Agilent 1100 MSD安捷伦科技有限公司)、自动过柱机(RF150美国电子特利丹公司)、真空油泵(DM2上海慕泓真空设备有限公司)、薄层层析硅胶板(安徽良臣硅源材料有限公司)、点样毛细管(0.3×100 mm上海新北玻璃仪器厂).
1.2 主要药品
2,7-二羟基萘(上海阿拉丁生化科技股份有限公司)、FeCl3(上海凌峰化学试剂有限公司)、异丙醇(上海实验试剂有限公司)、无水甲醇、无水甲苯、N,N-二甲基甲酰胺(上海药明康德新药开发有限公司)、98%H2SO4(永华化学股份有限公司)、对甲苯磺酸(药明康德(物流库存))、乙腈(永华化学股份有限公司)、乙酸乙酯、石油醚、饱和碳酸钾、无水硫酸钠(上海素元化工有限公司).
2 实验内容
图1 合成路线图Fig.1 Synthesis roadmap
通过大量文献查阅,本着以简洁、实用、易于扩展的原则,拟采用的合成路线如图1.
2.1 1-(2,7-二羟基-1-萘基)萘-2,7-二醇的合成
反应始投料5 g,经过柱层析纯化后得3.75 g产物,纯度97.23%,收率达到75.42%,随后将反应投料放大至20 g,反应过程如下:
反应操作:室温下,在1 000 mL三口瓶中加入FeCl3(20.25 g,124.87 mmol,7.23 mL,2 eq)和去离子水(300 mL),搅拌至完全溶解,然后将2,7-二羟基萘(10 g,62.43 mmol,1eq)溶于IPA(40 mL)的溶液缓慢加入到上述混合液中,油浴升温到40 ℃,在此温度下搅拌3 h,直到TLC(石油醚:乙酸乙酯=3∶1)显示原料2,7-二羟基萘完全消耗,反应停止后取样进行LCMS分析.
反应后处理:将反应混合物冷却至室温,用乙酸乙酯(300 mL)稀释并室温搅拌1 h,加入600 mL水,分别用300 mL乙酸乙酯萃取两次,合并的有机相用无水Na2SO4干燥,抽滤,滤液减压浓缩,得到12 g棕褐色粗品.
反应纯化:通过硅胶柱纯化得到纯品(洗脱剂为石油醚∶乙酸乙酯=5∶1~3∶1).
反应结果:通过LCMS和1H NMR进行结构分析确认得到中间体1-(2,7-二羟基-l-萘基)萘-2,7-二醇(7.6 g,23.59 mmol,75.56%产率,98.80%纯度,绿色固体).随后该反应按此条件放大到至160 g,经柱层析纯化得到125 g,纯度97.12%,收率达到78.35%.
2.2 1-(2,7-二羟基-1-萘基)-7-甲氧基--萘-2-醇的合成
反应投料:室温下,在500 mL干燥三口瓶中加入1-(2,7-二羟基-I-萘基)萘-2,7-二醇(7.6 g,23.88 mmol,1 eq)和甲醇(250 mL),室温下充分搅拌至原料完全溶解,然后再N2保护下缓慢滴加H2SO4(4.68 g,47.75 mmol,2.55 mL,2 eq),将反应混合物油浴升温至80 ℃,在此温度下搅拌回流72 h,直到TLC(石油醚∶乙酸乙酯=3∶1)指示起始原料完全消耗,反应停止后取样进行LCMS分析.
反应后处理:将反应混合物减压浓缩除去甲醇,然后用乙酸乙酯(500 mL)稀释并搅拌1 h,转移至1 000 mL烧杯,用饱和K2CO3溶液中和至pH≈7,加入1 000 mL水,并分别用500 mL乙酸乙酯萃取3次,合并的有机相用无水Na2SO4干燥,抽滤,滤液减压浓缩,得到8 g粗品
反应纯化:通过硅胶柱纯化得到纯品(洗脱剂为石油醚:乙酸乙酯=5∶1~2∶1)
反应结果:得到1-(2-羟基-7-甲氧基-1-萘基)-7-甲氧基-萘-2-醇(5 g,23.48 mmol,56.47%的产率,93.40%的纯度),为浅黄色固体.并将产物送至LCMS和1H NMR分析,谱图显示结构正确.随后该反应在此条件下分别放大至80 g和160 g,产率分别为62.25%和75.18%,放大后收率更高.
2.3 2,12-二甲氧基-7-氧代-[5]-螺旋烯的合成
反应投料:室温下,在500 mL单口瓶中加入I-(2-羟基-7-甲氧基-1-萘基)-7-甲氧基-萘-2-醇(5 g,14.44 mmol,1eq)和甲苯(200 mL),将反应油浴升温至140 ℃,搅拌至固体完全溶解,此温度下加入p-TsOH(3.73 g,21.65 mmol,1.5 eq),接着搅拌回流72 h,反应过程中及时分水,直到TLC(石油醚∶乙酸乙酯=3∶1)显示原料完全消耗,反应停止后取样监测即进行LCMS分析.
反应后处理:将反应混合物冷却至室温,减压浓缩,用乙酸乙酯(500 mL)稀释并搅拌1 h,饱和K2CO3溶液中和至pH≈7.加入1 000 mL的水,并分别用500 mL乙酸乙酯萃取3次,合并的有机相用无水Na2SO4干燥,抽滤,滤液减压浓缩,得到粗品6 g.
反应纯化:通过硅胶柱层析纯化得到纯品(洗脱剂为石油醚∶乙酸乙酯=5∶1~2∶1).
反应结果:通过LCMS和1H NMR进行结构分析确认得到2,12-二甲氧基-7-氧代-[5]-螺旋烯(1.5 g,4.42 mmol,产率30.63%,纯度96.80%,白色固体).随后该反应在此条件下分别放大至10 g和50 g,通过硅胶柱层析和打浆纯化产率分别达到45.21%和52.50%,放大收率更高.
3 结果分析与讨论
对各步合成的产物进行结构表征和性质探索.
3.1 1-(2,7-二羟基-1-萘基)萘-2,7-二醇谱图分析
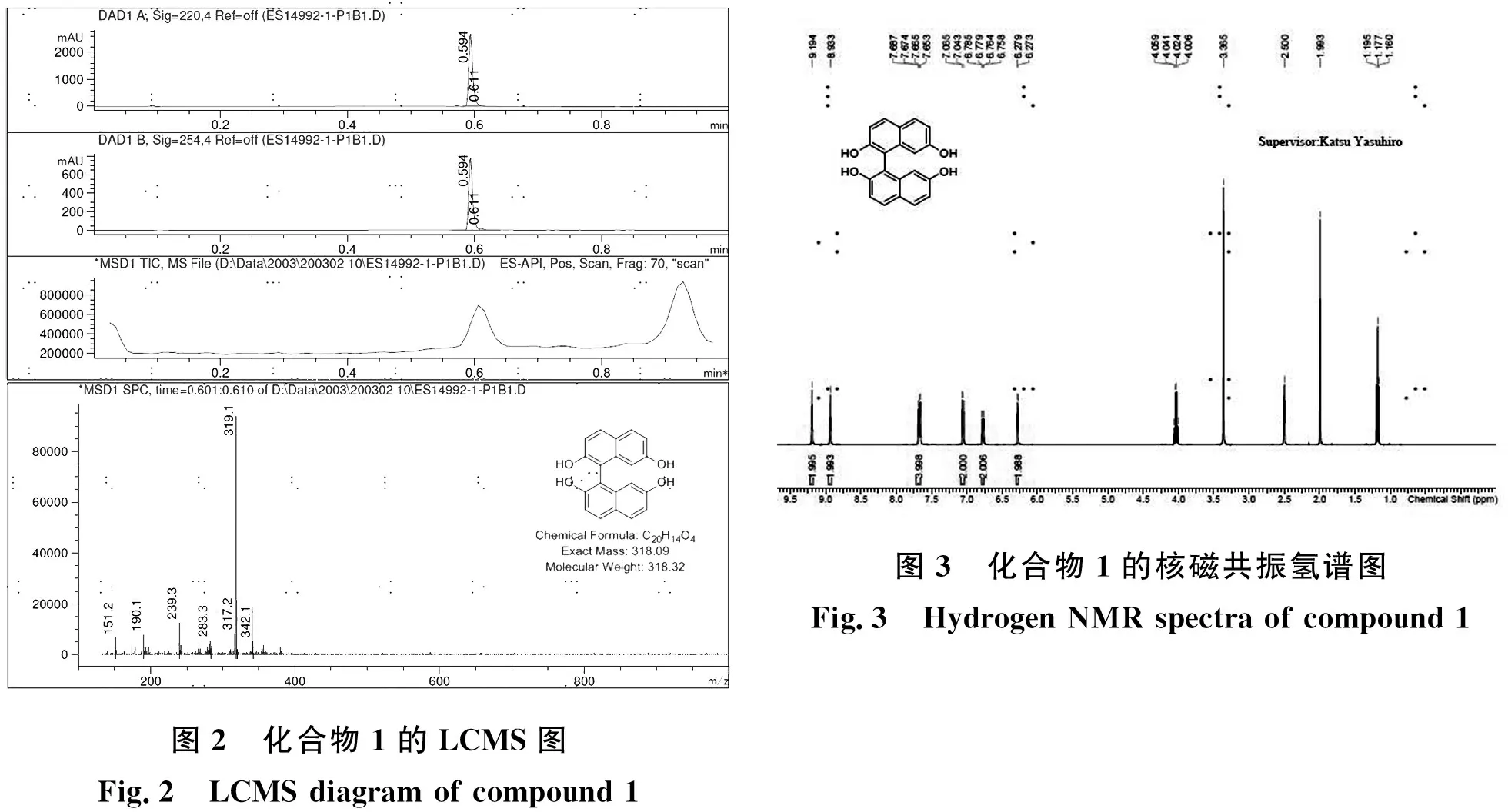
图2 化合物1的LCMS图Fig.2 LCMS diagram of compound 1图3 化合物1的核磁共振氢谱图Fig.3 Hydrogen NMR spectra of compound 1
(1) LCMS谱图
LCMS:tR=0.594 min in 5-95AB_1min_220&254_Agilent.M;Column:Agilent Poroshell 120 EC-C18 2.7um 3.0*30mm,MS(ESI)m/z [M+H]+=319.1
图2分析:由LCMS图可以看出,保留时间在t=0.594 min处,MS(ESI)m/z=319.1[M+H]+,与目标产物分子量相符.
(2)1H NMR
1H NMR:1H NMR(400 MHz,DMSO-d6)δ=9.19(s,2H),8.93(s,1H),7.67(dd,J=4.4,8.4 Hz,4H),7.05(d,J=8.4 Hz,2H),6.79-6.75(m,2H),6.28(d,J=2.4 Hz,2H).
图3分析:在核磁共振氢谱图上,化学位移2.50左右处是多重峰,为溶剂氘代DMSO;化学位移9.19以及8.93左右处有单峰,是四个羟基上的4个H;化学位移7.68-6.27左右为萘环上的10个H;化学位移1.17、1.99、4.02左右是乙酸乙酯的峰,说明有乙酸乙酯残留,但是可以除去,去除方法在后续结果讨论中提及;化学位移3.33左右是水峰.从谱图上可以看出,氢的个数和化学位移基本和化合物1吻合,结构正确.
3.2 1-(2,7-二羟基-1-萘基)-7-甲氧基--萘-2-醇的谱图分析
(1)LCMS图
LCMS:tR=0.715 min in 5-95AB_1min_220&254_Agilent.M;Column:Agilent Poroshell 120 EC-C18 2.7um 3.0*30mm,MS(ESI)m/z [M+H]+=347.1.
图4分析:由LCMS图可以看出,峰值在t=0.715 min时出现,化合物的相对分子质量为346.12.当tR=0.715时,LCMS的主峰值为347.1,和化合物2的相对分子质量吻合,是M+1,所以该峰值是目标产物.
(2)1H NMR图
1H NMR:1H NMR (400 MHz,DMSO-d6)δ=9.15(s,2H),7.77(dd,J=3.2,8.8 Hz,4H),7.16(d,J=8.8 Hz,2H),6.95(dd,J=2.4,8.8 Hz,2H),6.36(d,J=2.4 Hz,2H),3.46(s,6H)
图5分析:在核磁共振氢谱图上,化学位移2.50左右处是多重峰,为溶剂氘代DMSO;化学位移3.33左右为水峰;化学位移9.14左右处是单峰,为酚羟基上的2个氢;化学位移7.78-3.35左右为萘环上的10个氢;化学位移4.02左右是单峰,为甲氧基上的6个H.图上可以看出过柱后的产品比较纯,基本没有杂质和溶剂残留,氢的个数和位置与化合物2吻合,结构正确.
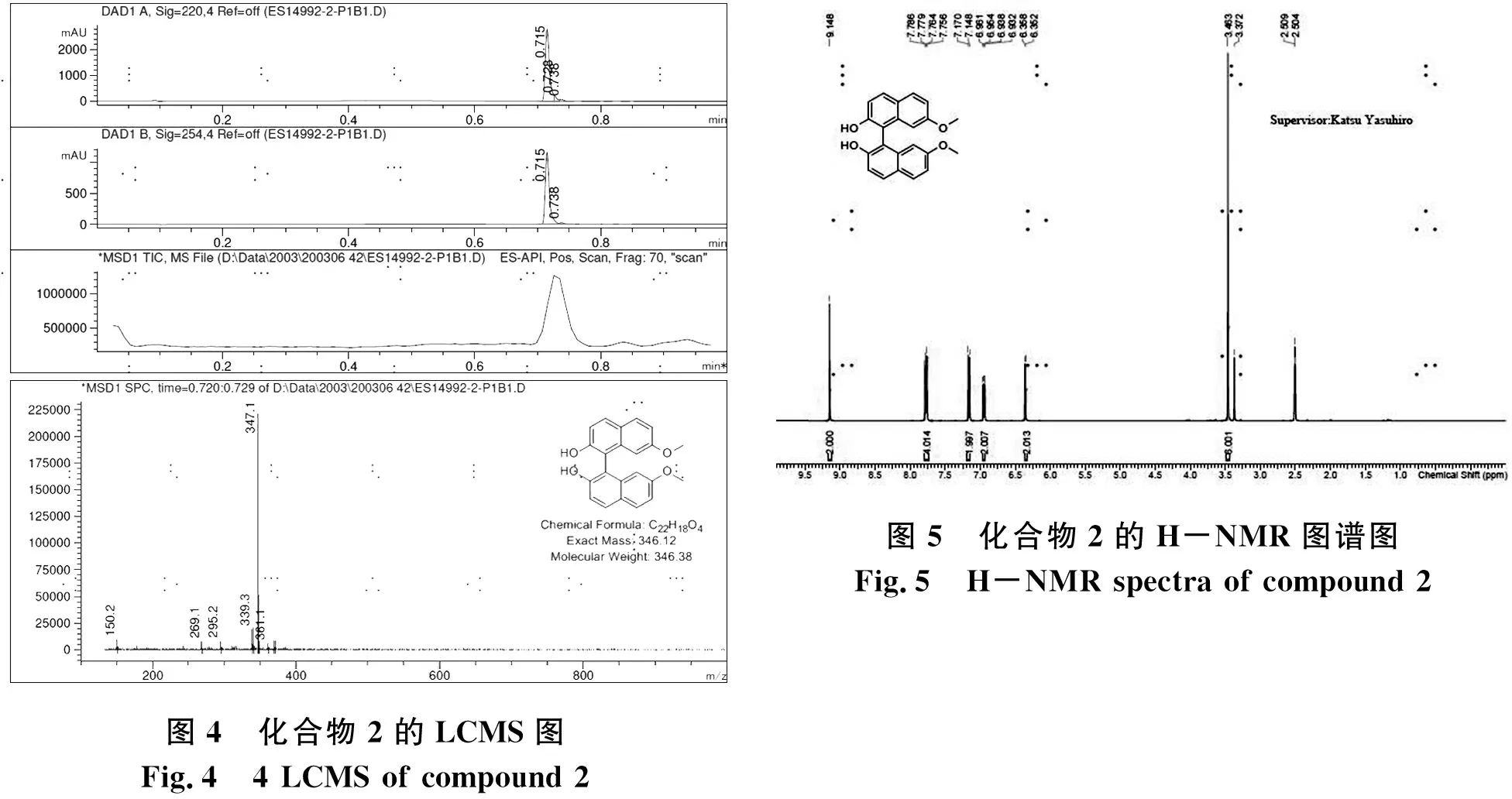
图4 化合物2的LCMS图Fig.4 4 LCMS of compound 2图5 化合物2的H-NMR图谱图Fig.5 H-NMR spectra of compound 2
3.3 2,12-二甲氧基-7-氧代-[5]-螺旋烯谱图分析
(1)LCMS图
LCMS:tR=0.926 min in 5-95AB_1min_220&254_Agilent.M;Column:Agilent Poroshell 120 EC-C18 2.7um 3.0*30mm,MS(ESI)m/z[M+H]+=329.1.
图3-6分析:由LCMS图可以看出,峰值在t=0.926 min时出现,化合物的相对分子质量为328.11.当tR=0.926时,LCMS的主峰值为329.1,和化合物3的相对分子质量吻合,是M+1,所以该峰值是最终产物的相对分子质量.
(2)1H NMR图
1H NMR:1H NMR(400 MHz,DMSO-d6)δ=8.39(d,J=2.0 Hz,2H),8.15(d,J=9.0 Hz,2H),
8.06(d,J=8.8 Hz,2H),7.84(d,J=8.8 Hz,2H),7.37(dd,J=2.3,9.0 Hz,2H),4.02(s,6H).
图3-7分析:在核磁共振氢谱图上,化学位移2.50左右处是多重峰,为溶剂氘代DMSO;化学位移3.33左右是水峰;化学位移8.38-7.37左右为萘环上的10个H,化学位移4.02左右是单峰,为甲氧基上的6个H,从图中看出,经纯化后目标物已经相对较纯,氢的位移和个数和目标物吻合,结构正确.

图6 目标物的LCMS图Fig.6 LCMS of the target product图7 目标物的H-NMR图Fig.7 H-NMR spectra of the target product
4 结果讨论
4.1 氧化偶联反应以及优化
这一步反应主要目的是生成联萘酚,本文采用的是异丙醇作溶剂,三氯化铁为催化剂进行合成,这一步合成反应进行相对顺利,难点是产物提纯,其优化过程如下:
(1)用乙腈∶水=1∶9打浆,56%的收率,97%的纯度,但是有乙腈残留,未达到预期要求;
(2)用乙腈∶正庚烷=1∶2打浆,杂质无法除去,未达到预期要求;
(3)纯水打浆,67%的收率,有乙腈残留,同样未达到预期要求;
(4)柱层析纯化后的产物粗品继续用乙腈溶解,60 ℃下减压浓缩除去乙酸乙酯,然后用纯水在室温下打浆过夜,真空抽滤,减压浓缩,最终得到纯品,纯度基本保持在99%以上,并且基本产品损失率很底.
4.2 醚化反应以及优化
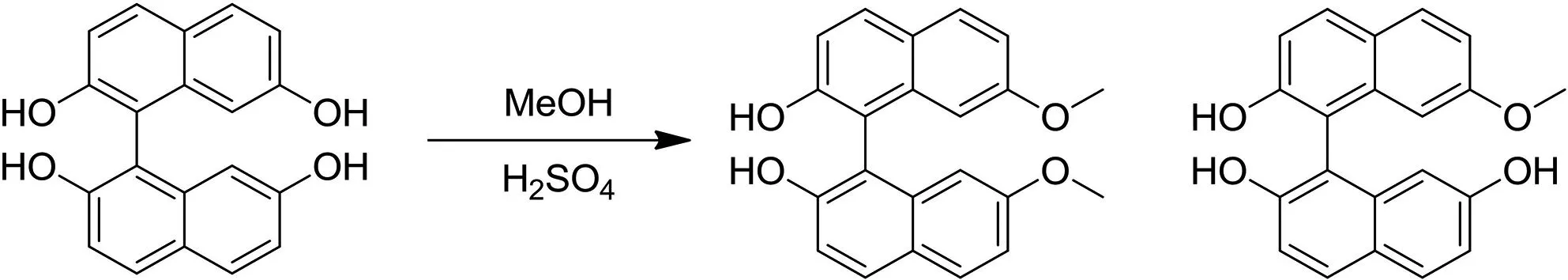
这一步反应的主要目的是保护酚羟基,本文采用的是甲基作为酚羟基的保护基,甲醇为溶剂,该反应最大的问题会产生副产物,但是经过不断改变硫酸当量数,这个问题也得到了一定解决,最终提高了产物的收率,而生成的副产物也能通过快速制备色谱进行初步分离.产物以及副产物结构式如下:
当加入2倍当量的硫酸,在80 ℃下反应72 h后LCMS监测到2.81%的1-(2,7-二羟基-1-萘基)-7-甲氧基-萘-2-醇和50.88%的副产物;当加入4倍当量的硫酸,在80 ℃下反应72 h后LCMS监测到28.53%的产物和47.74%的副产物,当加入10倍当量的硫酸,在80 ℃下反应72 h后LCMS监测到43.36%的产物和42.43%的副产物.可以看出,增加硫酸当量数能明显增加产物的收率,副产物的量也在逐渐减少.
这一步反应的后处理涉及萃取,因为产物不好溶,所以萃取的时候不容易分层,在后处理部分将反应液在搅拌下缓慢加入水里,会有固体析出,滤出的固体性状和纯度相对较好,可以直接用于下一步反应,不需要过柱纯化.
4.3 酸催化环化反应以及优化
该环化反应有水生成,需要及时将生成的水分离除去,才有利于反应正向进行,后处理如果将反应溶液在搅拌下缓慢倒入冰水中析出产物,但因为产物包夹的水分较多,会造成抽滤困难,费时费力,收率也会受到影响.本实验进行这改进即先用饱和K2CO3中和至pH≈7,随后往反应体系中加入适量水和大量的乙酸乙酯,萃取分液,将产物尽量萃取出来,然后减压浓缩,再进行下一步纯化.
为了能达到纯度要求,实验中尝试了多种纯化方法,除了用硅胶柱层析纯化,还尝试了打浆纯化的方法,分别用甲苯,正庚烷,乙腈,石油醚,乙酸乙酯打浆,最后用确定下了石油醚∶乙酸乙酯(3∶1)混合体系打浆,得到99%纯度的产物,并且有较高的收率.
5 结论和展望
5.1 结论
经过文献调研和试剂查询,设置合成路线,通过每一步的条件筛选和摸索,以及产品后处理、纯化和分离的工艺优化,克服了小试收率低,纯化困难,或产品纯度不高等各种问题,最终找到一条切实可靠的合成路线和工艺优化方法,概括起来主要在以下几个方面:
第一步的氧化偶联反应主要困难在纯化上,通过探索不同的纯化条件,最终找到一种理想方案:先将简单柱层析纯化后的粗品用乙腈溶解,通过减压浓缩将残留乙酸乙酯除去,然后用纯水打浆最终得到99%纯度的中间体.
第二步醚化反应难点在于有单个醚化副产物的生成,导致产物收率较低,通过改变H2SO4当量发现增加H2SO4的当量有利于提高产物收率,最终使用10倍当量的H2SO4,产物从2.8%增加到43.4%,对于该反应后续放大非常有利.
第三步酸催化反应难点在于后处理以及纯化,针对该类放大反应,用萃取的方法比直接抽滤相对简单,但柱层析纯化后纯度难达到要求.后通过用不同溶剂尝试打浆纯化,找出以石油醚∶乙酸乙酯=3∶1在20 ℃打浆2 h,可以得到纯度在98%以上的目标物,且收率较高.
5.2 展望
目前的合成路线和工艺优化还有一些值得改进和方向,主要在以下方面:
(1)路线能否缩短:整个合成路线一共涉及五个步骤,过程比较复杂,有些步骤反应时间较长,时间和成本的投入相对较高,目前少量文献上提及更短的合成方法,不过收率和纯度都不太理想,在这方面还有很大的探索空间.
(2)工艺能否放大:目前的规模在百克级左右,能否进步放大和重现收率和后处理纯化工艺有待进步研究.
(3)是否适合工业生产:实验室规模的小试、中试到工艺生产还有很长一段路要走,反应条件是否适合生产设备,后处理和纯化工艺是否能够套用,杂质的分离和鉴别等都需要进步的研究和优化.
猜你喜欢
今日农业(2020年20期)2020-12-15 15:53:19
能源(2018年10期)2018-12-08 08:02:48
铜仁学院学报(2018年6期)2018-07-05 09:47:36
中成药(2017年4期)2017-05-17 06:09:50
能源(2016年10期)2016-02-28 11:33:30
分析测试学报(2015年8期)2016-01-13 06:19:28
无机化学学报(2014年8期)2014-02-28 17:32:33
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:32
杭州师范大学学报(自然科学版)(2013年1期)2013-10-28 05:04:19
江西理工大学学报(2013年1期)2013-03-20 14:57:07
