
固体饮料中一种新型卡巴地那非类似物的定性鉴别和定量分析
2022-05-21 06:54:48夏金涛吴婉琴朱松松范志勇王会霞彭青枝
分析测试学报 2022年5期
夏金涛,吴婉琴,朱松松,江 丰*,范志勇,王会霞,彭青枝,杨 总
(1.湖北省食品质量安全监督检验研究院,湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430075;2.上海爱博才思分析仪器贸易有限公司,上海 200335)
随着社会的快速发展,人们工作生活压力的加大,勃起功能障碍(Erectile dysfunction,ED)成为当前发病率较高的疾病之一,从而使得抗ED 药物有着极大的市场需求。以西地那非为代表的选择性5型磷酸二酯酶抑制剂作为治疗男性抗ED 的特效药之一,在食品、保健食品中的非法添加时有报道[1-9]。对那非及其衍生物母核不同位置的取代基进行修饰改造,可得到数量众多的化学品[10-11]。这些未经《药品生产监督管理方法》[12](GMP)认证的结构类似物,若被掺入到食品、保健食品或中成药中,可能会对患者的健康和生命造成严重威胁。因此,建立多技术联用手段对新型、非法添加、结构未知的那非类似物进行定性鉴定和定量分析,对于保障广大民众“舌尖上的安全”具有重要意义。
目前,国家监管部门发布的那非类物质检验方法共有8 项[10-11,13],检测方法大多采用高效液相色谱-质谱联用法,涵盖100 多种那非衍生物。但一些不法商家为了规避执法人员的检查,尝试向保健食品中非法添加具有相似药效、检测标准以外的类似物,从中牟取暴利。近年来,丙氧苯基硫代艾地那非[8]、丙氧苯基西地那非[14]、丙氧苯基硫代羟基豪莫地那非[15]等新型那非衍生物被发现非法添加到食品及保健食品中。基于现行标准的局限性和被动性以及当前非法添加物筛查的需求,建立一种检测食品中非法添加新型那非类物质的通用、可行的技术手段显得十分有必要。
本研究运用超高效液相色谱-四极杆-飞行时间高分辨质谱(UPLC-Q-TOF MS)、超高效液相色谱-线性离子阱/静电场轨道阱高分辨质谱(UPLC-LTQ/Orbitrap MS)、核磁共振一维氢谱、碳谱和二维相关谱对固体饮料中非法添加那非类似物进行定性解析,最终确定该物质为N-苯基丙氧苯基卡巴地那非,并采用高效液相色谱-串联质谱(HPLC-MS/MS)进行定量分析。本方法通过多技术联用对新型非法添加卡巴地那非类似物进行了快速定性定量分析,为非法添加物质的筛查提供了必要的检测手段。
1 实验部分
1.1 试剂、材料与仪器
氘代二甲亚砜(DMSO - D6,D:99%)、氘代氯仿(CDCl3,D:98%)(Sigma - Aldrich 贸易有限公司);乙腈、甲醇(色谱纯,德国Merck 公司);甲酸(质谱纯,美国Fisher Scientific 公司);乙酸铵(色谱纯,Fisher Chemical 公司);超纯水(美国Millipore 公司);其他试剂均为分析纯,购于国药集团化学试剂公司;柱层析硅胶(300 ~400 目,青岛海洋化工厂分厂);薄层层析硅胶板(Silica gel 60 F254,德国Merck公司);5 mm核磁管(美国Norell公司);咖啡空白基质及固体饮料:电商平台。
液相色谱(Dionex Ultimate 3000)- 四极杆- 飞行时间质谱仪(SCIEX Triple TOF 5600 +)(美国SCIEX公司);Ultimate 3000高效液相色谱仪(配备二元泵、柱温箱、自动进样器)、线性离子阱/静电场轨道阱高分辨质谱仪(美国Thermo 公司);Bruker Avance III HD 600 MHz 超导傅里叶变换核磁共振波谱仪(德国Bruker 公司);Waters e2695 高效液相色谱仪(配备Waters 2998 光电二极管阵列检测器,美国Waters 公司);AB Sciex Qtrap 6500 型串联四极杆质谱仪(美国SCIEX 公司);HEI-VAP 旋转蒸发仪(德国Heidolph GmbH);Dryfast 2045 真空泵(德国Welch 公司);ZF-20D 暗箱式紫外分析仪(上海宝山顾村电光仪器厂);230 Volt 涡旋振荡器(美国Talboys 公司);S 180H 超声波清洗仪(德国艾尔玛公司);Centrifuge5810常温低速离心机(德国Eppendorf公司);XS204 电子天平(瑞士Mettler Toledo公司)。
1.2 定性分析条件
1.2.1 色谱条件TOF MS 色谱条件:色谱柱:Accucore aQ(150 mm × 2.1 mm,2.6 μm);柱温:35 ℃;流速:300 μL/min;进样量:5 μL;流动相:A为0.1%甲酸水溶液,B为甲醇。梯度洗脱程序:0.0~2.0 min,95% A;2.0~20.0 min,95%~5% A;20.0~25.0 min,5% A;25.0~26.0 min,5%~95%A;26.0~30.0 min,95%A。
Orbitrap MS色谱条件:色谱柱:Acquity UPLC BEH C18(2.1 mm×50 mm,1.7 μm),流动相:A为乙腈,B 为水(含0.1%甲酸)。梯度洗脱程序:0.0~3.0 min,1%A;3.0~5.0 min,1%~95%A;5.0~7.0 min,95%A;7.0~7.5 min,95%~1%A;7.5~10 min,1%A。流速:300 μL/min;柱温:35 ℃;进样量:5 μL。
1.2.2 质谱条件TOF MS 条件:离子源:电喷雾离子源(ESI 源);扫描方式:正离子模式;电喷雾电压(IS):5 500 V;气帘气压力(CUR):241 325 Pa;雾化气压力(GS1):241 325 Pa;辅助气压力(GS2):241 325 Pa;离子源温度(TEM):500 ℃;全扫描一级质谱(MS),质量采集范围100 ~1 000 Da;全扫描二级质谱(MS/MS),质量采集范围50 ~1 000 Da。
Orbitrap MS条件:离子源温度:325 ℃;离子传输杆温度:350 ℃;鞘气压力:275 800 Pa;辅助气压力:68 950 Pa;扫描模式:Full-scan ddMS3,一级优先扫描目标母离子m/z515.276 7,二级优先扫描目标子离子m/z161.107 3;一级扫描高能碰撞裂解(HCD)碎裂能量:40 eV,二级扫描HCD 碎裂能量:60 eV;一级扫描分辨率:R=60 000,二级扫描分辨率:R=30 000;一级扫描自动增益控制目标离子数(AGC):100 000,二级扫描自动增益控制目标离子数(AGC):10 000;质量偏差窗口:10 ppm。
1.2.3 核磁条件1H NMR 脉冲序列:zg30,采样次数(NS):4,谱宽(SW):20 ppm,观察道中心频率偏置(O1P):6.175 ppm;13C NMR脉冲序列:zgpg30,NS:500,SW:240 ppm,O1P:100 ppm。
1.2.4 液相色谱纯度测试条件色谱柱:C18色谱柱(250 mm ×2.1 mm,5.0 μm);柱温:30 ℃;流速:1 mL/min;进样量:10 μL;流动相:A 为甲醇,B 为0.02 mol/L 乙酸铵水溶液,采用二元等度洗脱(70%A、30%B),检测波长:230 nm。
1.3 HPLC-MS/MS定量分析条件
1.3.1 色谱条件色谱柱:C18色谱柱(2.1 mm×75 mm,2.1 μm);柱温:35 ℃;流速:300 μL/min;进样量:2 μL;流动相:A为0.1%甲酸水溶液,B为甲醇。梯度洗脱程序:0~1.0 min,95%A;1.0~4.0 min,95%~5%A;4.0~7.0 min,5%A;7.0~7.1 min,5%~95%A;7.1~10.0 min,95%A。
1.3.2 质谱条件离子源:ESI 源;检测方式:多反应离子监测(MRM);扫描方式:正离子模式;IS:5 500 V;CUR:310 275 Pa;GS1:241 325 Pa;GS2:241 325 Pa;TEM:500 ℃。
1.4 样品前处理
1.4.1 待测样品制备称取1 g(精确至0.001 g)待测样品置于50 mL 容量瓶中,加甲醇适量,涡旋振荡2 min 后,超声辅助提取10~15 min,冷却至室温,用甲醇定容,转移至50 mL 离心管中,3 900 r/min离心5 min,上清液过0.22 μm有机微孔滤膜。取续滤液,样品定性可直接上机测试,样品定量则根据实际浓度用甲醇水溶液(1∶1)稀释至线性范围内,备用。
1.4.2 核磁纯样制备称取约36.0 g 样品于500 mL 圆底烧瓶中,分3 次加入300 mL 甲醇,样品每次经涡旋振荡10 min,超声辅助提取10 min 后,抽滤。下层液体经旋蒸仪旋干后,通过快速柱层析硅胶对提取物进行净化,用暗箱式紫外分析仪对薄层层析硅胶板爬板结果进行初步判断,收集产品进行富集,旋干溶剂,用真空泵将样品抽干。取约10 mg 粉末,以DMSO-D6或CDCl3为溶剂,终产品进行核磁一维和二维谱测试。
1.5 标准溶液配制
1.5.1 标准储备液精密称取N-苯基丙氧苯基卡巴地那非10 mg,用甲醇溶解并稀释至50 mL,摇匀,制成质量浓度为200 μg/mL的标准储备液。于-20 ℃避光贮存,有效期为3个月。
1.5.2 标准工作液准确吸取标准储备液适量,用甲醇水溶液(1∶1)稀释,摇匀,作为系列标准工作液,质量浓度依次为2、5、10、20、50 ng/mL。
2 结果与讨论
2.1 定性分析
2.1.1 目标化合物的一级分子量扫描、二级及三级质谱裂解分析取待测样品,按照“1.2.1”的TOF MS 色谱条件进行分离,发现该化合物的保留时间为15.41 min(图1A),随后按“1.2.2”的TOF MS 质谱条件直接进样进行一级分子量扫描,得到准分子离子峰为m/z515.276 7([M+H]+),经拟合后确定化合物的分子式为C29H34N6O3(质量偏差0.4 ppm)(图1B)。进一步以m/z515.276 7为母离子进行子离子扫描,得到二级碎片质谱图(图2A),该化合物具有明显的西地那非特征离子m/z311.113 0、283.118 3、166.097 0、147.007 7,说明其至少具有图2A的母核结构。由二级碎片离子m/z353.159 0可推测,从m/z353.159 0到311.113 0丢失了C3H7,但不能确定C3H7在化合物中的连接方式。从m/z515.275 1到353.159 0可能丢失了C10H13N2,为进一步确定其基本结构,按“1.2.1”和“1.2.2”,采用UPLC-LTQ/OrbitrapMS对其进行了三级碎裂(图2B)。可见此部分具有明显的苯环(m/z77.038 43)、丢失1 个亚甲基或氮原子(m/z146.083 82)的结构。
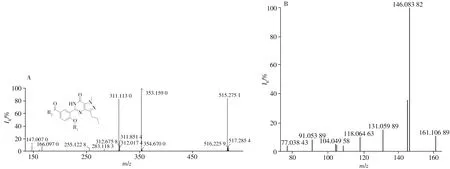
图2 目标化合物的二级碎片质谱图、母核结构(A)及三级碎片质谱图(B)Fig.2 Secondary fragment MS spectrum,parent nucleus(A)and third fragment MS spectrum(B)of desired product
2.1.2 目标化合物的核磁谱图分析取“1.4.2”的纯化样品约10 mg,以DMSO - D6为溶剂,按“1.2.3”条件测试样品的氢谱(图3A)和碳谱(图3B),得到相关数据如下:1H NMR(600 MHz,DMSO)δ12.01(s,1H),7.74(d,J=2.2 Hz,1H),7.59(dd,J=8.5,2.2 Hz,1H),7.26 ~7.19(m,3H),6.95(d,J=8.1 Hz,2H),6.8(t,J=7.2 Hz,1H),4.15(s,3H),4.08(t,J=6.3 Hz,2H),3.67(brs,4H),3.34(s,3H),3.18(brs,4H),2.76(t,J=7.5 Hz,2H),1.78 ~1.6(m,4H),0.96(t,J= 7.4 Hz,3H),0.91(t,J= 7.4 Hz,3H);13C NMR(151 MHz,DMSO)δ168.8,158.0,154.1,151.3, 149.3, 145.4, 138.4, 131.5, 130.3, 129.4, 127.9, 124.7, 123.1, 119.9, 116.4,113.0,70.5,49.0(overlapping),38.3,27.7,22.4,22.2,14.3,10.9。
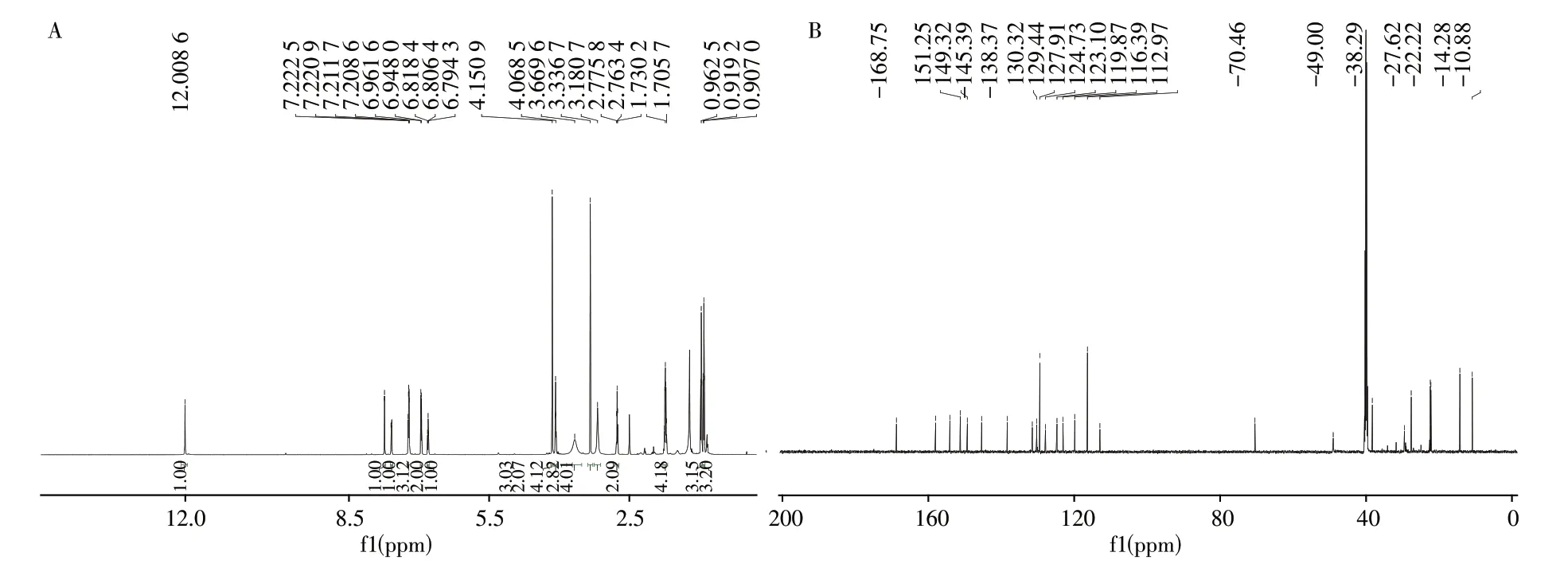
图3 目标化合物的1H NMR谱(A)和13C NMR谱(B)Fig.3 1H NMR(A)and13C NMR(B)spectra of desired product
通过1H NMR 观察到氢原子总数为34 个,与“2.1.1”质谱分析所得分子式(C29H34N6O3)结果一致;通过脂肪区氢原子的裂分形式4.08(t,J= 6.3 Hz,2H)ppm,1.78 ~1.68(m,4H)ppm,0.96(t,J=7.4 Hz,3H)ppm 可以确证图2中R1为正丙基,且有1个亚甲基信号与吡唑环上丙基的亚甲基信号重叠。二维TOCSY核磁谱显示芳香区的氢原子属于同一自旋体系,结合13C NMR 129.4 ppm,116.4 ppm 推测该自旋体系为1 个三取代和1 个单取代苯环,根据芳香区氢原子的数目确定图2 中R2含有单取代苯环。根据13C NMR 168.8 ppm,判断三取代苯环上连有酰胺键。脂肪区的化学位移3.67(brs,4H),3.18(brs,4H)ppm 说明R2含有2 对对称的亚甲基结构。结合文献[16]比对,确定该化合物为N-苯基丙氧苯基卡巴地那非(图4),该结构与1H NMR、13C NMR 及Dept-90/135-13C分析高度吻合。氢谱、碳谱归属如表1。
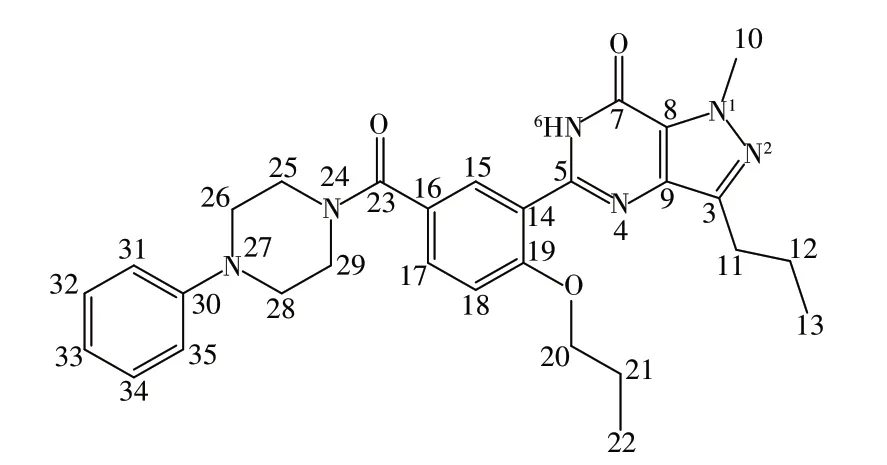
图4 N-苯基丙氧苯基卡巴地那非的结构Fig.4 Structure of N-phenylpropoxyphenyl carbodenafil
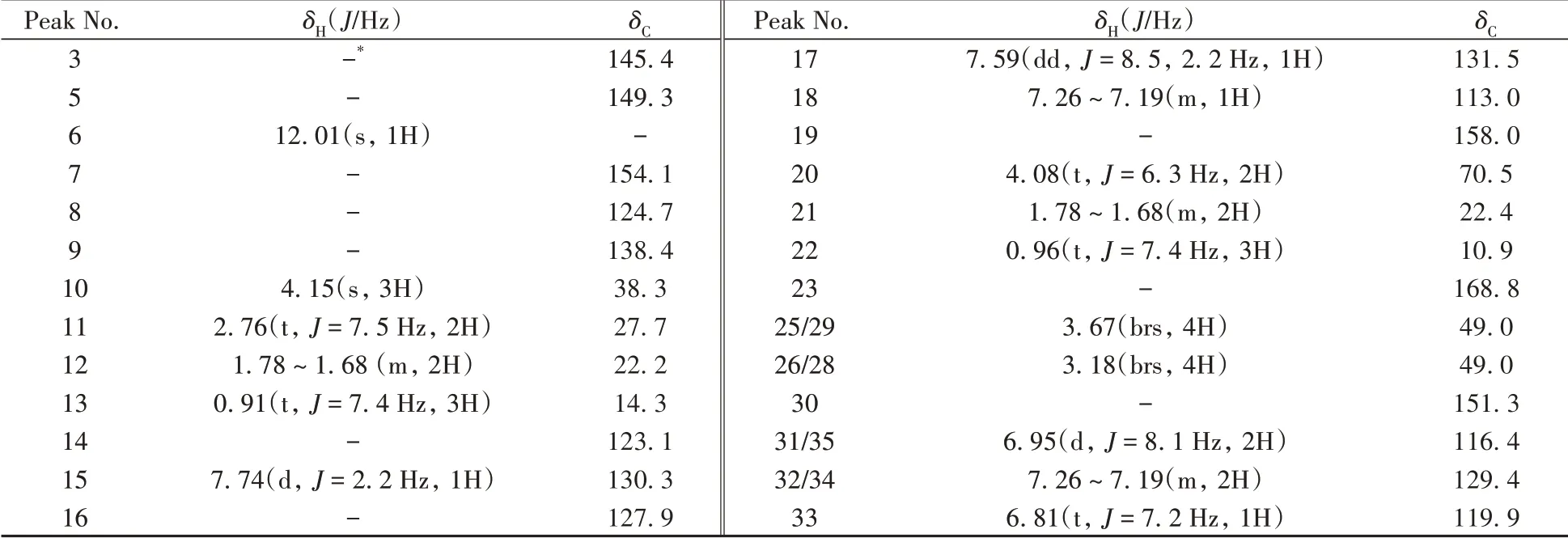
表1 N-苯基丙氧苯基卡巴地那非的核磁共振数据归属(DMSO-D6)Table 1 NMR data of N-phenylpropoxyphenyl carbodenafil(DMSO-D6)
根据确认的结构,结合质谱分析,归纳整个分子的裂解途径如图5所示。在溶液中N-苯基丙氧苯基卡巴地那非(分子式C29H34N6O3,分子量514.269 2)以游离态形式存在,正离子模式下,m/z为515.276 7。初次裂解得到m/z为353.160 8 和161.107 3 的特征碎片离子峰,随后2 个特征碎片峰分别发生进一步裂解。其中m/z为353.160 8 的碎片离子在高电压下发生正丙基断裂,得到m/z为283.119 0 的碎片峰,随后进一步脱羰基得到m/z为255.124 0 的碎片离子。m/z为161.107 3 的碎片经Orbitrap 三级裂解分析,分别得到m/z146.096 4 和77.038 6的特征离子峰。
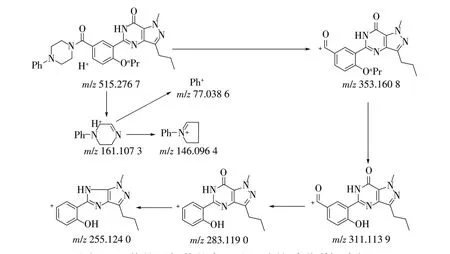
图5 N-苯基丙氧苯基卡巴地那非的质谱裂解途径Fig.5 MS fragment pathway of N-phenylpropoxyphenyl carbodenafil
2.2 定量分析
2.2.1 目标化合物的纯度评价取待测样品,按“1.2.4”条件测得目标化合物N-苯基丙氧苯基卡巴地那非的最大吸收波长为204 nm,纯度为99.78%。
2.2.2 基质效应采用相对响应值法对咖啡基质的基质效应(ME)进行考察,计算公式如下:ME =(k2/k1-1)×100%。式中:k1为标准溶液曲线的斜率;k2为基质标准曲线的斜率。一般来说,当基质效应为-20%~20%时,表明基质效应在可接受范围内;反之则应考虑基质效应对实际检测的影响。实验发现,咖啡空白基质在原液配制标准曲线中存在一定基质效应,而采用甲醇水溶液(1∶1)稀释10倍的空白基质原液配制标准曲线,基质效应有所减弱,进一步采用甲醇水溶液(1∶1)稀释100 倍的空白基质原液配制标准曲线,基质效应均在-20%~20%之间。因此,本实验中阳性样品基质在前处理中增加稀释过程,以降低或消除基质效应,并采用空白溶剂标准曲线进行定量。
2.2.3 线性关系、检出限与定量下限取“1.5.2”制备的标准工作液,按照“1.4.1”和“1.3”方法进行前处理及测定,采用外标法定量。以目标化合物的峰面积为纵坐标(y),质量浓度为横坐标(x)制作标准曲线,得到目标化合物在2~50 ng/mL 质量浓度范围内呈良好的线性关系,线性方程为y=358 084x-695 617,相关系数(r2)为0.999 1。通过测定不同加标水平的样品溶液,以3 倍信噪比确定方法检出限(LOD),以10 倍信噪比确定方法定量下限(LOQ),并综合考虑与补充检验方法BJS201805[13]的一致性,得到咖啡样品中N-苯基丙氧苯基卡巴地那非的LOD 为0.05 mg/kg,LOQ 为0.1 mg/kg。
2.2.4 回收率与相对标准偏差对咖啡空白基质样品进行三水平加标回收率实验,分别制成加标水平为0.1、0.2、1.0 mg/kg 的样品,每个浓度做6 次平行实验,考察方法的回收率和相对标准偏差(RSD)。得到不同加标水平下的平均回收率为89.2%~93.1%,RSD 为1.8%~3.1%,可以满足分析要求。
2.3 实际样品的测定
采用本方法对电商平台购买的10批次固体饮料进行测定。结果表明,有5批次样品检出N-苯基丙氧苯基卡巴地那非,检出含量分别为214、218、229、268、880 mg/kg。
3 结 论
本研究建立了一种液相色谱-质谱联用、核磁共振表征的多检测技术手段鉴定固体饮料中新型、非法添加、结构未知的卡巴地那非类似物—N-苯基丙氧苯基卡巴地那非的方法。该化合物目前无CAS 号登记,其毒理数据也未见报道。HPLC-MS/MS定量分析表明,N-苯基丙氧苯基卡巴地那非的线性范围为2~50 ng/mL,相关系数(r2)为0.999 1,方法的平均回收率为89.2%~93.1%,RSD 为1.8%~3.1%。对市场上10批次咖啡固体饮料进行检测,发现5批次样品检出该非法添加物,含量为214~880 mg/kg,说明该物质在固体饮料中的非法添加存在潜在的安全隐患。
猜你喜欢
山西文学(2023年6期)2023-06-09 09:10:42
食品安全导刊(2021年20期)2021-08-30 06:39:48
农药科学与管理(2019年8期)2019-11-23 08:04:44
当代化工研究(2016年5期)2016-03-20 16:21:35
色谱(2015年6期)2015-12-26 01:57:30
化学工业与工程(2015年1期)2015-02-10 03:01:33
特产研究(2014年4期)2014-04-10 12:54:22
Sciences in Cold and Arid Regions(2014年6期)2014-03-31 00:28:31
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年7期)2014-02-28 17:32:28
