
我国污染场地化学氧化修复技术应用特征及再利用潜在腐蚀风险分析
2022-05-19 12:26王珍霞宋久浩苑文仪吴乃瑾李培中
环境科学研究 2022年5期
王珍霞,宋久浩,苑文仪,李 翔,吴乃瑾,李 艺,李培中*
1. 上海第二工业大学资源与环境工程学院,上海 2012092. 北京市科学技术研究院资源环境研究所,北京 1000953. 工业场地污染与修复北京市重点实验室,北京 100095
长期工业生产活动所产生的水相、气态和固体等不同环境介质的污染,大部分最终汇存、富集于土壤和地下水中[1-2]. 据报道显示,我国污染场地面积超过1×104m2的数量在50×104块以上,其中大部分集中在长江三角洲、珠江三角洲和东北老工业基地等重点区域,以氯代烃、苯系物和多环芳烃类等有机物为主[3]. 有机物污染会对人体健康、饮用水安全和生态环境等产生威胁,可能引发后果较为严重的环境污染事件[4-6]. 国内有机污染场地常用的修复技术包括热脱附、生物修复和化学氧化等,与其他技术相比,化学氧化具有节能、效率高、周期短等优势,是近年来发展迅速的污染场地修复技术之一. 氧化反应需要相对较好的传质介质,与非饱和土壤相比,地下水具有更好的电子传递条件,因此化学氧化修复更多地应用于受污染土壤和地下水的协同修复.
常用的化学氧化试剂包括高锰酸盐、Fenton或类Fenton、臭氧和过硫酸盐等,这些氧化试剂具有非常强的氧化势能,并且部分药剂持续作用时间较长[7-8].美国超级基金场地等国外典型污染场地修复后,主要再开发为绿化、公园等非高强度建设用地,一般会进行长达几年或十几年的氧化副反应自然恢复过程,因此针对修复后场地直接开发利用所产生的残余腐蚀风险研究还相对较少[9-11]. 我国污染场地修复后主要用于城市建设用地,达标后主要用于住宅和公共服务等建设强度较大的项目. 由于修复周期非常短,且修复标准相对国外更为严格,修复过程中通常存在氧化试剂的过量使用[12]. 氧化试剂反应过程中会产生较多的硫酸根离子、氯离子等盐分副产物,当这些盐分浓度过高时,会对再开发利用过程的混凝土、金属地基等造成盐分腐蚀风险[13-14]. 因此,化学氧化修复技术在实际工程应用过程中或修复完成后,经常会受到环境、建筑设计等领域研究和管理人员的质疑,在一定程度上限制了该技术规模化推广应用[15-16].
化学氧化修复会产生残余副产物,造成土壤和地下水环境条件改变. 这一现象已经引起研究人员的关注,但系统性研究还相对较少,尤其是在次生腐蚀风险方面[11,17-18]. 鉴于此,该文首先梳理了近年来我国化学氧化修复技术的应用进展及行业特征;然后,针对应用相对较多的过硫酸盐氧化法可能产生的氧化剂残留、副产物及其他环境条件等潜在的次生问题,进行了综合分析;最后,结合资料文献分析和国内外案例场地调研信息,对这些环境变量可能产生的次生腐蚀风险机理和腐蚀风险程度进行讨论分析,探索建立化学氧化与次生腐蚀之间的关联性,以期为化学氧化修复技术的持续、健康发展提供科学数据支撑.
1 我国有机污染场地化学氧化修复技术的应用特征
从国外地下水污染修复技术发展趋势来看,化学氧化技术是近年来主流的修复技术之一. 1982—2017年,美国超级基金场地污染地下水原位修复技术统计案例720个,其中化学氧化技术占比达23%,是仅次于生物修复技术的修复技术[4]. 随着欧盟水资源管理政策日趋严格,在对受农药污染水体修复技术的统计过程中也发现,高级氧化技术占比相对较高,高达30%[19]. Wacławek等[20]通过对水和废水研究文献统计分析发现,自2007年以来,以过硫酸盐为代表的高级氧化技术研究快速增加. 根据中国环境保护产业协会的统计情况来看,化学氧化修复技术已从10年前的不足1%快速增加到目前的20%左右(见图1),其中原位化学氧化技术占比相对较多. 根据问卷调查和网络检索,收集到国内近期137个场地污染修复案例,发现我国污染场地化学氧化修复技术的应用有4个特征:主要应用于中小型污染地块、水土协同修复且介质复杂、过硫酸盐占比过大且副产物较多、修复周期短且修复药剂过量现象严重.
图1 我国化学氧化修复技术的工程化应用发展概况Fig.1 The general situation of engineering application of chemical oxidation remediation technology in China
1.1 主要应用于中小型污染地块
目前,化学氧化技术主要的修复对象为毒性强、有异味、具有土壤与地下水交叉污染等特点的中小型有机污染场地. 这些场地亟需修复后再开发为住宅等高附加值的敏感用地,因此需要采取较为彻底的降解修复技术,如化学氧化、焚烧、热脱附等. 焚烧、热脱附等修复技术虽然修复效果较好,但是装备复杂、投资成本高、二次污染控制严格,相对较高的综合修复单价成本是中小型污染场地难以承担的. 化学氧化对修复药剂要求较高,但设备简单、施工准备快,且二次污染较易控制,综合修复单价成本相对较低,对于这些中小型污染场地适用性更强,尤其是异味控制较为敏感的场地[3]. 另外,中小型场地总体修复费用预算较低,很难引入大型、有经验的修复公司进行修复施工,中小型修复施工单位所占比例更大,进而可能造成后期修复的准确性和科学性存在更多的不确定性.
1.2 水土协同修复且介质复杂
从国内实际化学修复工程案例的分布情况可以看出,现有修复场地主要分布在我国中东部沿海、沿江等经济发达地区. 这些地区地下水水位埋深相对较浅,有机污染物通常会造成土壤和地下水复合污染,需要对土壤和地下水进行协同修复,相应地,原位化学氧化修复技术应用案例要远多于相对干燥的北方土壤[21-22]. 原位化学氧化对前期污染精准调查和修复施工单位经验要求相对较高,加上这些沿海、沿江区域水文地质条件相对较为复杂,因此导致外源注入修复药剂与目标污染物有效接触和充分反应存在很多的制约因素. 大多数场地修复效果通常很难一次直接达到预期目标,经常发生污染反弹现象,甚至造成重复修复施工的情况.
1.3 过硫酸盐占比过大且副产物较多
从氧化剂种类来看,美国实际应用中高锰酸盐的累积使用占比最多,为33.8%,双氧水次之,达到26.4%[23],从时间的角度来看,早期是以高锰酸盐为主,后来Fenton或类Fenton(双氧水)及过硫酸盐才逐步得以应用[24]. 与美国的发展情况不同,高锰酸盐和双氧水分别由于颜色、安全管制等原因在国内的推广应用较少,过硫酸盐应用相对更多. 过硫酸盐作为近年来备受关注的新型高级氧化试剂:①具有相对更高的氧化势能,E0(SO4—·/SO4
2—)=2.60~3.10 V(以氢电压计);②具有更广泛的活化方法;③具有更宽泛的氧化反应条件;④具有更低廉的综合成本;⑤具有更持久的作用时间[25]. 对2012年以来在互联网上公开的修复工程效果评估(验收)报告统计分析发现,25个原位化学氧化修复场地中有17个使用过硫酸盐作为氧化剂. 过硫酸盐施用过程中需要进行活化,实际工程中主要活化方法有过渡族金属离子活化和碱活化,但两种活化方式均会引起环境pH变化,并且造成硫酸盐、还原硫等副产物大量存在.
过硫酸盐是一种持久性较强的氧化剂,不同环境介质中其滞留时间存在差异(见图2),在未被污染的含水层介质中半衰期为2~600 d,在地下水中的滞留时间大于5个月[26]. Lemaire等[27]研究了原位化学氧化处理多环芳烃污染土壤,发现处理4 d后仍有30%~45%的过硫酸盐存在. 相比于·OH,过硫酸盐产生的SO4

图2 过硫酸盐在不同环境介质中滞留时间的对比Fig.2 The comparison of residence time of persulfate in different environmental media
—·对氧化底物的选择性更强,也更加稳定[28],在环境中的半衰期更长,残存期间可能对环境造成更大的影响.
1.4 修复周期短且药剂过量现象严重
我国实际污染场地修复后主要用于城市建设用地,达标后大部分场地短期内均用于住宅和公共服务等建设强度较大的项目,其要求修复周期尽可能缩短,且短期修复标准相对国外更为严格,因此造成了修复过程中化学氧化试剂通常过量使用[12]. 研究表明,化学氧化剂与目标污染物的最佳配比为20∶1(摩尔比)[29],但是由于土壤中自身含有较多的有机质等还原组分,要想达到完全去除污染物的目的,实际修复工程中化学氧化剂的添加量通常要远大于理论值,最大配比能够达到100∶1(摩尔比)以上[11]. 国外对修复案例场地跟踪调查发现,原位化学氧化对污染物的去除率仅为51%~72%,并且78%的场地会发生污染反弹的现象[30]. 而国内已经进行修复效果评估的场地中,绝大多数在2~3个月内均能达到预期的修复目标,说明实际化学氧化试剂的添加量可能远超过实际需求量.
同时由于工程应用经验相对缺乏,导致药剂注入方式、混合方式和药剂用量等实际工程应用参数还处于探索阶段,很多实际修复工程中化学氧化剂的添加量达到了3%~5%(相对土壤质量比). 例如,调研发现,山东某污染场地原位化学氧化使用1%~5%(相对土壤质量比)过硫酸盐修复后,土壤中残留的硫酸盐含量高达5400 mg/kg,是背景值(85.6 mg/kg)的63倍以上,远超过GB 50021—2001(2009年版)《岩土工程勘察规范》中对建筑基础(混凝土结构)土壤的腐蚀标准(2250 mg/kg),相应的地下水硫酸盐也严重超标(见图3).

图3 某污染场地原位化学氧化后土壤和地下水中硫酸盐浓度的变化情况Fig.3 The Changes of sulfate concentration in soil and groundwater after in-situ chemical oxidation in a contaminated site
2 过硫酸盐修复后场地的潜在腐蚀风险
过硫酸盐激活产生强氧化性的硫酸根自由基(SO4—·),可以将目标有机污染物破坏去除,但是过量的氧化剂在土壤中持续发生氧化作用,可能对后期开发利用中地基、管道等地下构筑物表面产生较强的腐蚀作用. 外加的过硫酸盐导致短期pH降低,增加酸 碱 腐 蚀 作 用[31],以 及 副 产 物SO42—等 盐 分 浓 度 升高[32],潜在盐分腐蚀作用增强[13,33];同时,地下环境条件改变可能导致硫酸盐还原菌等微生物的异常生长,造成潜在的生物腐蚀. 在上述多重腐蚀的共同作用下,可能对过硫酸盐修复后场地再开发利用中地下构筑物产生明显的负面影响.
2.1 氧化剂腐蚀
一般情况下非活化过硫酸盐的半衰期为2~600 d,氧化剂残留量与投放量呈正相关,投放120 d后,10%浓度的过硫酸盐仍存在40%左右[34]. 过硫酸盐溶液对铁制品具有较大的腐蚀性,且腐蚀作用随着浓度升高而增强,10%浓度的溶液作用一周可造成10.09%的减重[12]. 外加药剂、加热变性[35]和其他综合作用造成土壤和地下水pH发生较大改变(见图4),同时H+参与阳极电化学反应〔见式(1)~(3)〕[36],加速Fe等金属的腐蚀损耗,较低的土壤pH(pH≤3)产生的腐蚀作用比其他盐分腐蚀作用更强.

图4 不同投加药剂对反应体系pH的影响[40]Fig.4 The influence of different agents on pH of reaction system[40]

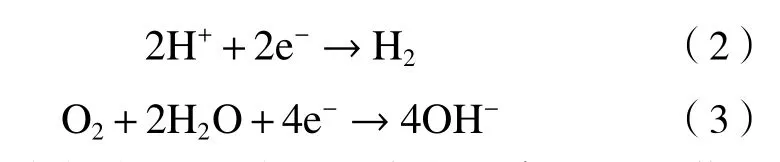
酸性土壤与中性或碱性土壤中的腐蚀有显著差异,酸性厌氧环境会影响管线钢材的腐蚀[37],在酸性土壤中作为阴极去极化剂的铁氧化物会刺激碳钢的腐蚀过程[38]. 另外,过硫酸盐的过量投加会造成土壤酸化,修复过程中生成大量SO42—和H+,在酸性条件下结合即形成腐蚀性硫酸,对建筑地基造成一定的腐蚀风险[39].
碱活化能够缓解过硫酸盐处理后反应体系酸化的问题,但碱活化体系中对pH的控制要求较高,依靠强碱的作用将pH保持在较高范围,甚至维持零级碱度. 根据近期国内污染场地过硫酸盐化学氧化修复的应用情况(见表1),发现使用碱活化后,多数场地都存在pH升高现象,造成土壤碱化. 碱性、潮湿的环境下,Fe等金属会发生吸氧腐蚀[41].

表1 近期国内污染场地的过硫酸盐化学氧化修复应用和pH变化Table 1 The recent application of persulfate chemical oxidation and change of pH in contaminated sites in China
2.2 盐分腐蚀
大量注入土壤和地下水中的过硫酸盐导致地下残留的硫酸盐等副产物浓度大幅升高(部分场地地下水中最大浓度超过20000 mg/L)[45],这种现象往往会持续时间较长,过高的盐分浓度通常会对混凝土、金属地基等地下构筑物基础产生盐分腐蚀风险. 研究[48]表明,混凝土在硫酸盐侵蚀下的劣化是从有害离子通过水的输送开始的,SO42—通过水的扩散作用渗透到硬化的水泥浆中,基质的许多成分易受SO42—的影响.通过室内试验模拟不同硫酸盐溶液浓度和干湿循环条件下混凝土的腐蚀情况,结果表明,土壤中外加的盐分对构筑物具有较强的腐蚀作用(见表2). 影响混凝土侵蚀程度的因素较多,包括混凝土自身的材质、暴露方式、外加盐分的种类和浓度等.
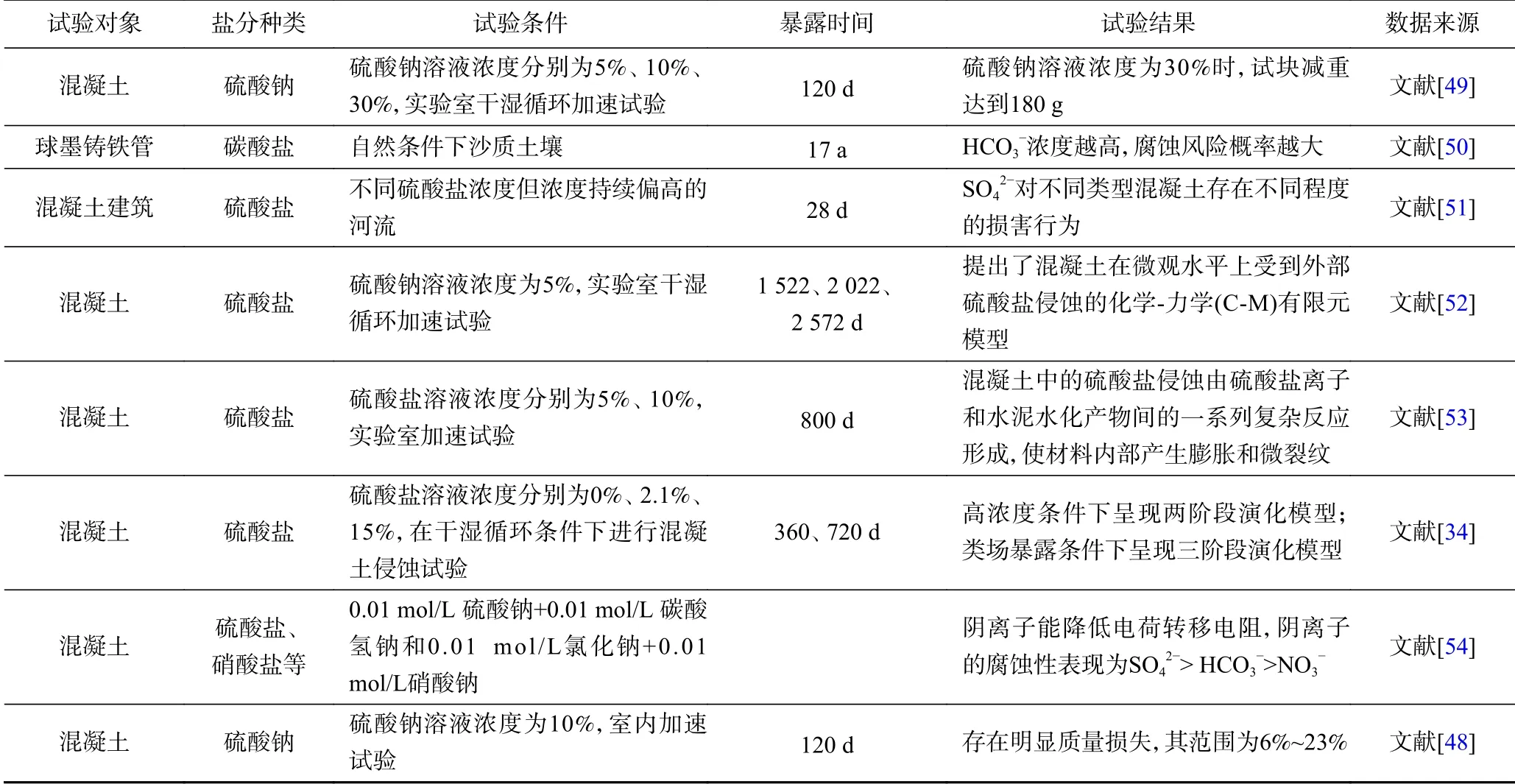
表2 基于盐分对混凝土侵蚀模拟试验的情况总结Table 2 The summary of the simulation test of concrete corrosion by salt
混凝土在低浓度的硫酸钠溶液中,质量、膨胀和抗压强度呈现出三阶段演化模型,而在高浓度的硫酸钠溶液中呈现两阶段演化模型[34]. SEM和EDX分析表明,变质混凝土样品中除钙矾石和石膏[55]外,还有大量的盐结晶(见图5). 早期增强阶段,归因于硫酸盐反应产物(如石膏和钙矾石)在混凝土内部的细化和充填作用;中期孵化阶段,混凝土内部的大量小孔被填充,该阶段是混凝土损坏的累积过程;退化阶段,混凝土内部的膨胀压力超过混凝土的抗拉强度造成混凝土出现开裂,导致渗透率增加,对混凝土结构影响非常严重,危及整个建筑物的可靠性[53].

图5 混凝土腐蚀产物SEM显微图像和腐蚀产物EDX光谱分析[34,48]Fig.5 SEM microscopic images of concrete and EDX spectroscopic analysis of corrosion products[34,48]
2.3 微生物腐蚀
据不完全统计,微生物腐蚀约占我国腐蚀总量的20%[56-57]. 过硫酸盐原位化学修复过程中,地下深层环境条件的改变通常会影响土著微生物的群落结构,1%~5%过硫酸盐注入地下后,会导致微生物总量和多样性的减少,但硫酸盐还原菌(SRB)等异常微生物群落的快速生长,可能会对混凝土中硫酸盐的降解产生加速作用. 传统的腐蚀理论认为,微生物并未直接参与到腐蚀过程中,而是微生物的代谢产物产生腐蚀作用. 近年来随着研究的不断深入,发现微生物腐蚀作用是微生物直接或间接地参与了金属材料的腐蚀过程[58]. 总体来看,微生物腐蚀机理包括微生物代谢产物间接腐蚀、微生物直接腐蚀和氧浓差电池腐蚀.
微生物的新陈代谢过程通常会产生一些有机酸或无机酸等酸性腐蚀物质,这些物质会对地下构筑物产生腐蚀作用,进而形成微生物间接腐蚀作用. SRB可以利用SO42—作为电子受体,将其还原为S2—,通过自养生物反应生成硫化氢(H2S)、FeS或其他腐蚀性含硫物质[59]. Li等[60]通过长期的腐蚀过程监测,发现高浓度的H2S能够引起混凝土下水道的快速腐蚀,同样对碳钢也会造成严重的腐蚀作用[55,61]. 此外,高浓度硫酸盐环境条件下扩增的SRB,不仅可以利用还可以利用土壤溶液中的Fe(OH)2作为电子受体获得生长所需的能量,直接或间接地加速缝隙中钢的腐蚀[62-63]. 近年来的研究认为,微生物可以直接参与腐蚀反应,地下环境中大量存在的SRB直接从铁中获取电子及能量[64],加剧金属表面腐蚀,并形成具有半导体性质的矿壳产物[65]. 微生物还可以通过表面富集和繁殖形成生物被膜,从而对金属材料造成电化学腐蚀(见图6)[59]. 微生物及其代谢产物等在金属表面附着,形成厚度和分布不均的生物被膜[62],膜内具有复杂的电化学和生物电化学环境. 代谢活动产生的胞外多聚物,不仅阻碍了环境中氧气向金属材料表面迁移以及腐蚀产物的局部堆积,还促进了氧浓差电池的形成[59]. 氧浓差电池的形成使材料的局部腐蚀成为可能,胞外电子的传递加速,促进了微生物腐蚀过程[66-67].

图6 金属表面微生物腐蚀的电子传递方式[59]Fig.6 The electron transfer mode of microbial corrosion on the metal surface[59]
综上,过硫酸盐原位化学氧化在快速氧化去除目标有机污染物的同时,也可能对地下环境的氧化还原条件、盐分和微生物环境发生短期、中期和长期的影响,进而产生持续性的腐蚀作用,可能对后期开发利用中地基、管道等地下构筑物表面产生较强的腐蚀作用. 不同腐蚀阶段的作用机理和腐蚀速率存在一定的差异,总体上可以划分为3个不同的阶段(见图7):第1阶段为氧化剂腐蚀,该阶段腐蚀速率较快;第2阶段为盐分腐蚀,腐蚀速率相较于第1阶段变缓,会出现短期质量上升的现象;第3阶段为微生物与盐分腐蚀共同作用.
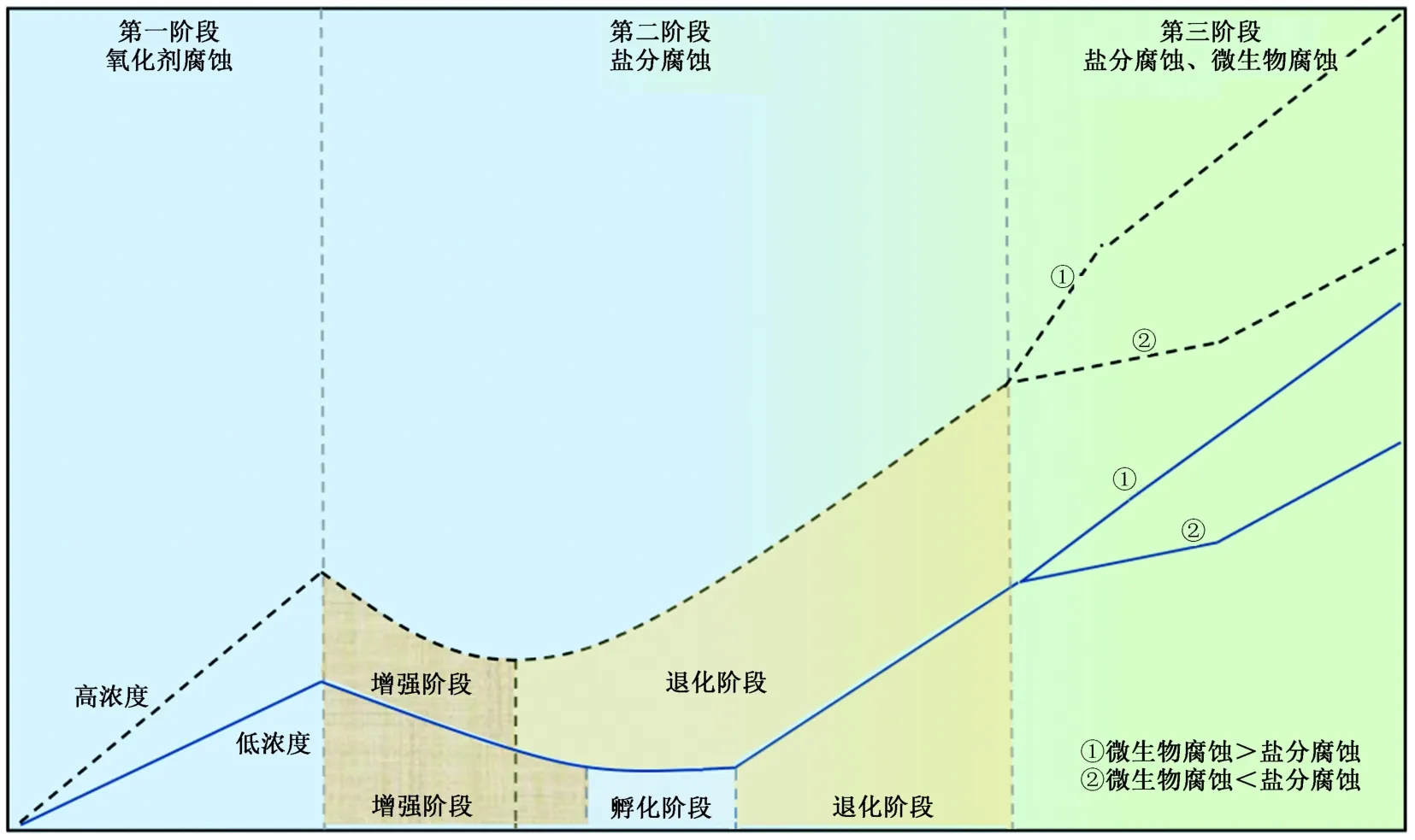
图7 不同腐蚀阶段的作用机理及腐蚀速率[12,34]Fig.7 The mechanism of action and corrosion rate in different corrosion stages[12,34]
3 结论与展望
a) 有机物污染土壤和地下水修复过程中,可能会由于外加药剂、加热变性和其他综合作用,造成土壤和地下水pH、氧化还原电位等发生较大改变,还可能出现副产物硫酸盐累积超标等问题,对混凝土、金属地基等工程基础产生盐分腐蚀风险,同时氧化剂本身和微生物群落也会强化潜在的腐蚀风险.
b) 在实际工程应用中要注意过硫酸盐的用量控制在一定的限度内,并尽可能少量多次注入,避免因药剂短期使用量过大而产生的氧化剂残留、酸碱副反应和次生腐蚀作用等实际工程问题,同时应该开展系统性的小试和中试研究.
c) 目前,针对修复后场地的腐蚀风险监测与评价方法的研究还相对较少,主要通过借鉴建筑领域、地下管网工程领域土壤和地下水腐蚀监测及等级划分的方法,探索构建工程腐蚀安全风险效果测量与评价体系,重点包括针对盐分、pH、氧化还原电位等影响因子的长期定量变化监测,以及其变化阈值对应的定性风险判断,最终形成有效的工程安全风险控制技术体系.
猜你喜欢
河南科技(2022年8期)2022-05-31
哈尔滨工业大学学报(2022年5期)2022-04-19
中国土壤与肥料(2021年5期)2021-12-02
建材发展导向(2021年15期)2021-11-05
绿色科技(2021年16期)2021-09-09
土壤与作物(2021年1期)2021-03-07
中学化学(2019年4期)2019-08-06
中学化学(2019年4期)2019-08-06
祝您健康·文摘版(2019年3期)2019-06-11
读者·校园版(2016年6期)2016-03-07
