
牛场辣椒的全基因组SNP标记分析
2022-05-16 08:58:02黄冬福何建文江叶莎付文婷范高领詹永发石燕金王楠艺
种子 2022年4期
黄冬福, 何建文, 江叶莎, 付文婷, 范高领, 吴 迪, 詹永发, 石燕金, 王楠艺
(1.贵州省农业科学院辣椒研究所, 贵阳 550009; 2.遵义市农业农村局, 贵州 遵义 563000)
辣椒为茄科辣椒属植物,有极大的利用价值,营养价值高可鲜食,是一种重要的调味品,富含的辣椒碱具有一定的药用价值,辣椒红素可用于食品及化妆品的着色。贵州六枝特区的牛场辣椒于2014年被认定为国家地理标志产品,具有果色深红,肉厚,辣味适中,香气浓郁等优异品质[1]。辣椒基因组上含有多种分子标记。在众多的分子标记中,SNP(单核苷酸多态性)最重要且最具吸引力,其高水平的多态性、共显性、高通量、丰富的信息量[2]广泛用于作物育种中的遗传多样性分析、基因组关联分析及比较基因组学[3-6]。
国内外研究者获得了辣椒的大量SNP标记。Siddique等[7]对188个辣椒重组自交系个体和352个辣椒材料进行重测序,开发了666 405个SNP标记,结合经典的QTL定位和全基因组关联分析,获得了3个赋予疫病广谱抗性的主效QTL。Wu等[8]对287个辣椒材料进行重测序,获得了9 557 790个SNP,通过全基因组关联分析,发现调控26个辣椒农艺性状的2 126个候选基因。Wang等[9]对辣椒不育系和保持系的线粒体基因组测序,获得了两者间的112个SNP,结合已知的CMS(细胞质雄性不育)基因特征,筛选出2个最有可能决定CMS的ORF。Han等[10]对208份辣椒材料进行重测序,开发了109 610个SNP标记,发现99个SNP与辣椒素显著关联。孙茜[11]对辣椒抗感黄瓜花叶病毒(CMV)的基因池进行重测序,获得了51 969 152个SNP标记,结合关联分析和经典的QTL定位,发现了抗CMV的1个主效QTL和2个微效QTL。赵红[12]对349份国内辣椒核心种质进行重测序,平均每份种质获得了7 425 498个SNP,通过全基因组关联分析,发现94个SNP与果实辣味等20个性状显著关联。
就目前的研究来看,各研究者开发了大量辣椒SNP标记,但是所用品种没有涉及牛场辣椒,而利用已有的SNP标记重新筛选牛场辣椒特异的SNP费时费力且无法保证数量与质量。另外,各研究者开发SNP采用的是简化基因组测序法,基于此法开发的SNP无法覆盖全基因组,SNP的数量及密度远不如全基因组测序法。因此,本研究利用全基因组重测序分析牛场辣椒的SNP标记,为牛场辣椒遗传图谱构建、重要农艺性状基因挖掘、遗传改良、品种鉴定与保护奠定基础。
1 材料与方法
1.1 材 料
牛场辣椒(CapsicumannuumL.),2014年被农业部认定为地理标志农产品,由贵州省辣椒研究所保存。取苗期的叶片用于全基因组重测序。
1.2 基因组DNA提取与DNA质量检测
采用DNA secure Plant Kit(TIANGEN)试剂盒提取基因组DNA;1%琼脂糖凝胶电泳检测DNA是否降解和污染;NanoPhotometer®spectrophotometer (IMPLEN, CA, USA)检查 DNA 纯度;使用2.0 Flurometer (Life Technologies, CA, USA)检测DNA 浓度。
1.3 测序文库构建
分别取检测合格的DNA 样品700 ng,通过Covaris破碎机打断成长度为350 bp的片段,使用NEB Next®Ultra DNA Library Prep Kit(NEB, USA)构建文库,并将index codes添加到每个测序样本中。使用AMPure XP系统(Beckman Coulter,Beverly,USA)纯化DNA,DNA片段的3′末端腺苷酸化后,连接具有发夹环结构的NEB下游衔接子以准备杂交;PCR反应前使用电泳来选择指定长度的DNA片段,在USER酶(NEB,USA)的作用下,37 ℃ 15 min、95 ℃ 5 min;然后用Phusion High-Fidelity DNA聚合酶、Universal PCR引物和Index(X)引物进行PCR, PCR产物用AMPure XP系统进行纯化。文库构建完成后,先使用Qubit 2.0软件进行初步定量,稀释文库至1 ng/μL,随后使用Agilent Bioanalyzer 2100软件对文库的insert size进行检测,insert size符合预期后,使用Q-PCR方法对文库的有效浓度进行准确定量(文库有效浓度>2 nmol/L),保证文库质量。
1.4 全基因组测序和质量控制
利用Illumina HiSeq 2000平台进行全基因组测序,测序生成的原始图像数据文件经碱基识别转化为原始测序序列(Illumina pipeline CASAVA v 1.8.2),然后通过质量控制去除以下不能用的reads,且两端reads均去除:
1) 带文库构建接头的reads;
2) 未知碱基超过10%的reads;
3) 低质量碱基(测序质量值≤5)超过50%的reads。
1.5 序列比对
质控后的有效测序数据经BWA软件比对到参考基因组(https://www.ncbi.nlm.nih.gov/genome/10896,v1.0),比对结果经SAMTOOLS和PICARD软件(http://picard.sourceforge.net)去除重复。
1.6 变异检测及注释
设定参数(-q 1-C 50-m 2-F 0.002-d 1000),用samtools软件检测原始的SNP,然后用如下标准进行过滤:
1) 变异位置的测序深度>4;
2) 质量值>20。
用ANNOVAR软件对SNP进行变异注释。
2 结果与分析
2.1 测序数据产出及与参考基因组比对分析
由表1可知,经过全基因组重测序,共获得785 408 260条Raw reads,平均读长150 bp,碱基总长为117.8 Gb;通过质量控制去除带接头的reads、未知碱基超过10%的reads以及低质量(Q值≤5)碱基数超过50%的reads,得到高质量的clean reads;clean reads共有783 349 390条,覆盖98.23%的基因组;去除clean reads中的非特异reads,获得771 948 477条有效reads,其中758 748 158条reads能锚定到“遵辣1号”参考基因组上,配对率为98.29%,平均测序深度为36.35×。

表1 牛场辣椒测序数据产出及与参考基因组比对情况Table 1 The sequencing data of Niuchang pepper and the result compared with the reference genome
2.2 染色体上SNP的分布规律和特征
根据与参考基因组的比较,SNP分为纯合和杂合类型,分别占SNP总数的59.12%、40.88%。每条染色体上的SNP总数不同,10号染色体上SNP总数最多(1 371 387个),4号染色体上SNP总数最少(349 894个)。每条染色体上纯合和杂合SNP的数量也不同, 10号染色体上的纯合SNP最多(889 193个),9号染色体上的纯合SNP最少(169 010个),9号染色体上的杂合SNP最多(779 326个),4号染色体上的杂合SNP最少(93 614个),具体见表2。
每条染色体上密度最高区域SNP出现频率不同,10号染色体上密度最高区域SNP出现频率最高(1 904个/100 kb),4号染色体上密度最高区域SNP出现频率最低(1 162个/100 kb),具体见表3和图1。

表2 每条染色体上SNP的数量Table 2 The number of SNP on each chromosome

表3 每条染色体上SNP密度最高区域及其出现频率Table 3 The frequency of SNP in highest SNP densityarea of chromosomes
2.3 基因组不同位置SNP分布特征
牛场辣椒基因组中的SNP分布在5个不同位置:基因上游、基因内、基因下游、基因上游/下游、基因间。(基因上游是指基因上游1 kb区域;基因内指基因内部;基因下游指基因下游1 kb区域;基因上游/下游指基因上游1 kb区域,同时也在另一基因的下游1 kb区域;基因间指两个基因间区)。5个不同位置的SNP数量不同且差异显著,SNP数量从多到少依次为基因间>基因内>基因上游>基因下游>基因上游/下游(表4),基因间、基因内、基因上游、基因下游的SNP占比依次为94.68%、3.64%、0.9%、0.74%(图2)。
基因内不同位置所含SNP数量也不同,基因内包含外显子、内含子、剪接位点3个位置,所含SNP数量分别为51 242、281 002、288个,SNP数量从多到少依次为内含子、外显子、剪接位点。针对外显子区域,根据SNP变异引起的密码子变化, 可将SNP分为4种类型:终止子获得、终止子缺失、同义突变、非同义突变,数量分别为710、188、19 079和31 265,SNP数量依次为非同义突变>同义突变>终止子获得>终止子缺失。

图1 每条染色体上的SNP密度热图 Fig.1 The density heat map of SNP on each chromosome
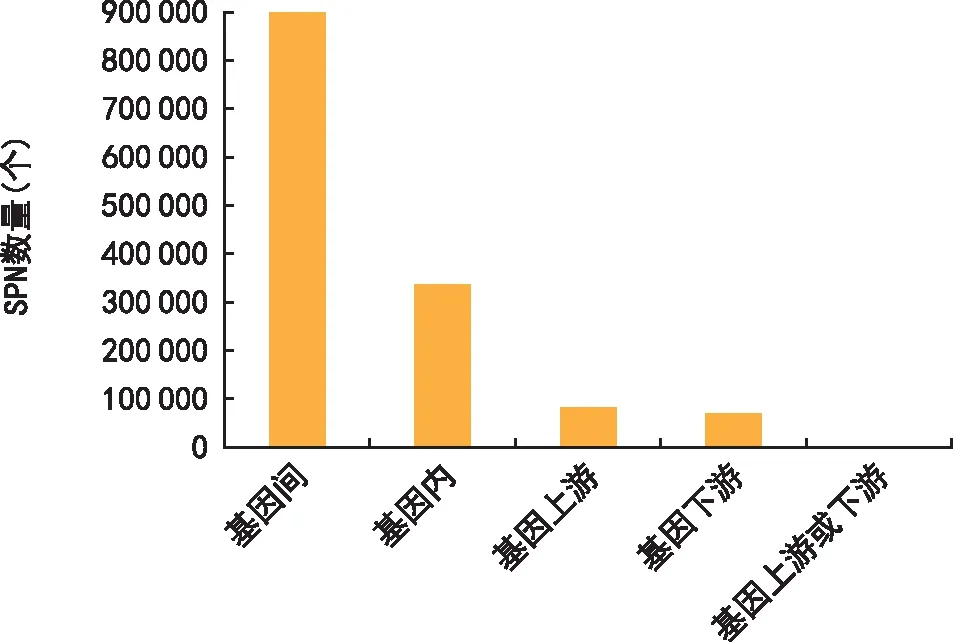
图2 牛场辣椒基因组中不同位置的SNP数量差异Fig.2 The number difference of SNP on the different position in the genome of Niuchang pepper

表4 牛场辣椒基因组中SNP的位置及相应数量Table 4 The position and corresponding number of SNP in the genome of Niuchang pepper
2.4 SNP突变频谱
全基因组SNP突变可分为6类:T∶A>G∶C,T∶A>C∶G,T∶A>A∶T,C∶G>T∶A,C∶G>G∶C和C∶G>A∶T。以T∶A>C∶G为例,此种类型SNP突变包括T>C和A>G。由于测序数据既可比对到参考基因组的正链,也可比对到参考基因组的负链,当T>C类型突变出现在参考基因组正链上,A>G类型突变即在参考基因组负链的相同位置,所以将T>C和A>G划分成一类。C∶G>T∶A的数量最多(3 109 688个),C∶G>G∶C的数量最少(474 542个)。T∶A>G∶C、T∶A>A∶T、C∶G>G∶C、C∶G>A∶T为颠换,总数为3 057 408个,T∶A>C∶G及C∶G>T∶A为转换,总数为6 094 214个,发生转换的数量是颠换的1.99倍(图3)。

图3 SNP突变频谱Fig.3 The mutation frequency and type of SNP
3 讨论与结论
3.1 基于WGRS技术开发辣椒SNP标记的高效性
辣椒全基因组序列的公布为其分子育种带来前所未有的机遇。想要开展辣椒分子育种,就必须对群体中所有个体进行基因分型。利用传统方法对辣椒进行基因分型费用高、耗时耗力,低水平的分子标记也是基因分型的重要挑战。基于新一代测序技术(NGS)的基因分型通量高、成本低、分子标记密度高。
全基因组重测序(WGRS)是新一代测序技术(NGS)的一种。利用WGRS技术开发分子标记具有标记密度大、有效标记多、准确率和特异性高、稳定性好的优势。本研究利用WGRS技术共鉴定出9 141 358个SNP,SNP的出现频率为1个/366 bp,其中51 242个SNP位于外显子。Ahn利用WGRS技术获得了6 840 889个辣椒SNP,其中39 955个SNP位于外显子[13]。
相比而言,利用简化基因组测序开发SNP标记的效率较低。Nimmakayala等[14]利用简化基因组测序仅获得66 960个辣椒SNP,SNP的出现频率为1个/40.7 kb,其中仅有2 521个SNP位于外显子。Nimmakayala等[15]采用简化基因组测序获得77 407个辣椒SNP,SNP的出现频率为1个/35.6 kb,其中26 697个SNP位于外显子。由此可见,利用WGRS获得的SNP标记数量、出现频率、外显子上的SNP数量远高于简化基因组重测序[14-15]。
功能标记是分子标记的一种,基于功能基因内的多态序列开发,与常规的分子标记相比,与表型的连锁程度更紧密[16],外显子上的SNP最可能作为功能标记。因此,通过检测功能标记能更准确地预测表型,在加速育种进程的同时极大地提高了标记辅助选择的准确性。本研究获得的51 242个外显子SNP作为潜在的功能标记,将成为辣椒分子育种的有力武器。
3.2 牛场辣椒SNP标记分布特征
SNP在牛场辣椒基因组上的分布表明,基因间的SNP数量比基因内多,是基因内的26倍,内含子区域的SNP数量比外显子多,是外显子区域的5.5倍,这种分布规律与Kim等[17]的研究结果相似。外显子上,SNP引起的终止子获得有710处,终止子获得是指碱基突变导致终止密码子提前出现。其产生截短的蛋白质,从而使基因散失原来的功能,并进一步引发作物表型变异。因此,终止子获得对基因功能研究具有重要意义。AFS1基因发生5 bp缺失及G/A转换,翻译提前终止,导致水稻小穗异常,小穗上多出一个外稃状器官,內稃发生不同程度的退化[18]。GmSG基因发生A/G转换,翻译提前终止,导致大豆种皮颜色由黄色转变为黄/绿色[19]。SNFL1基因单内含子上最后一个碱基发生单核苷酸突变,翻译提前终止,导致水稻旗叶变短变窄[20]。OsCUL 3 a蛋白翻译提前终止,导致水稻flg 22、几丁质诱导的活性氧、发病相关基因的表达量明显增加,进而产生类病斑[21]。
本研究发现,牛场辣椒10号染色体上的SNP数量最多,且18.17%的纯合SNP位于该染色体上,4号染色体上的SNP数量最少,与Nimmakayala[15]的研究结果不同,Nimmakayala的研究显示辣椒3号染色体上SNP数量最多,9号染色体上SNP数量最少,与Ahn[13]的研究结果也不同,Ahn的研究显示辣椒品种PRH 11号染色体上SNP数量最多,8号染色体上SNP数量最少。造成不同染色体上SNP分布频率差异的原因可能是不同研究者使用的辣椒品种不同。
SNP引起的点突变可分为转换和颠换。现有研究表明,人类基因组CpG中的胞嘧啶C能自发脱氨基变成胸腺嘧啶T,导致C∶G>T∶A的转换比T∶A>C∶G的转换多,同时,转换比颠换更容易发生,且转换的SNP是颠换的2倍[22]。牛场辣椒C∶G>T∶A的转换比T∶A>C∶G的转换多,且转换的SNP数量明显高于颠换,是颠换的1.99倍,与前人的研究结果基本一致。
本研究对牛场辣椒进行全基因组重测序,获得了9 141 358个SNP,SNP的出现频率为1个/366 bp,展示出WGRS技术开发辣椒SNP标记的高效性;其中51 242个SNP位于外显子上,外显子上的SNP具有开发成功能标记的巨大潜力,功能标记与表型的连锁程度更紧密,能极大地提高标记辅助选择的准确性,是辣椒分子育种的有力武器;外显子上的SNP产生了710处终止子,终止子获得会产生截短的蛋白质,使基因散失原来的功能,并进一步引发作物表型变异,对基因功能研究具有重要意义。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
辽河(2022年1期)2022-02-14 05:15:04
现代畜牧科技(2021年4期)2021-07-21 06:13:10
中国生殖健康(2020年4期)2021-01-18 02:58:10
科学之谜(2019年3期)2019-03-28 10:29:44
中国生殖健康(2018年4期)2018-11-06 07:12:16
科学之谜(2018年8期)2018-09-29 11:06:46
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
兽医导刊(2016年6期)2016-05-17 03:50:20
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
