
法布里病一例
2022-05-14 02:59赵梓纲赵纪敏周亚光
实用皮肤病学杂志 2022年1期
于 水,赵梓纲,赵纪敏,周亚光,周 勇
法布里病(Fabry disease)又称Anderson-Fabry综合征,由两位皮肤科医生威廉·安德森和约翰内斯·法布里于1898年首次描述[1]。此病是X染色体连锁遗传的α-半乳糖苷酶缺乏性疾病,致病基因定位于Xq22,编码了蛋白α-半乳糖苷酶A(α-GalA),皮肤主要表现为弥漫分布的血管角皮瘤。本文报道1例以皮肤血管角皮瘤、少汗、腹泻20余年为主诉的患者。
临床资料
患者,男,28岁。因全身紫红色斑丘疹,双侧手足感觉异常、触痛及少汗22年,于2017年6月就诊。22年前,患者腰部和膝部出现散在针尖大小、紫红色丘疹及斑丘疹,同时双手足出现阵发性僵冷、指尖触痛,躯干及手足少汗,伴无规律腹泻等症状,丘疹逐渐蔓延至胸部、臀部、阴囊及手指。患者父亲既往体健,母亲有系统性红斑狼疮病史,家族无其他类似患者。系统查体未见明显异常。皮肤科情况:全身多发簇集性紫红色针尖至粟粒大小斑丘疹,表面略粗糙,压之无褪色(图1);口腔黏膜无异常。背部丘疹组织病理示:表皮部分萎缩伴基底层色素颗粒增多,真皮浅层局灶血管扩张、充血,真皮下方纤维组织增生(图2)。眼底及听力检测未见异常。实验室及辅助检查:血浆α-半乳糖苷酶0.1 nmol/(h·mgPr)[正常值 29.0~ 64.4 nmol/(h·mgPr)],β-半乳糖苷酶 189.7 nmol/(h·mgPr)[88~ 220 nmol/(h·mgPr)];颅脑磁共振成像提示双侧顶叶皮层下可见小的点状缺血灶(图3);肌电图提示左侧上下肢皮肤交感反应测定波幅降低,上肢潜伏期延长;胃肠道内窥镜检查提示慢性非萎缩性胃炎。心脏、腹部、四肢血管超声多谱勒检查均无异常。在患者签署送检同意书的情况下,采集静脉血 2 ml,置于含乙二胺四乙酸(ethylenediamine tetraacetic acid,EDTA)抗凝试管中,送往北京康旭医学检验所进行Fabrydisease Panel检测。基因检测示患者 GLA基因核苷酸变异c.779G>C(编码区第 779 号核苷酸由 G 突变为 C)导致第 260 号氨基酸由甘氨酸变为丙氨酸(p.Gly260Ala)。该错义突变可能导致蛋白质功能受到影响,与法布里病相关。诊断:法布里病。建议患者父母进行基因检测,但因其父母无症状,未检。治疗:嘱患者避免劳累和剧烈运动,针对患者腹泻症状采取口服益生菌等对症处理;定期复查尿蛋白、颅脑磁共振等,并进行遗传咨询。截止发稿时止,已随访3年,患者病情无明显进展。
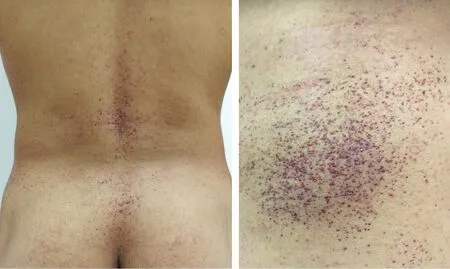
图1 法布里病患者背部皮损
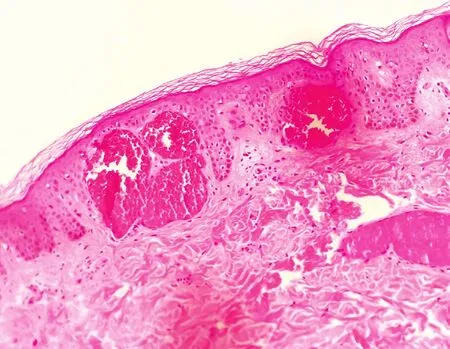
图2 法布里病患者背部皮损组织病理
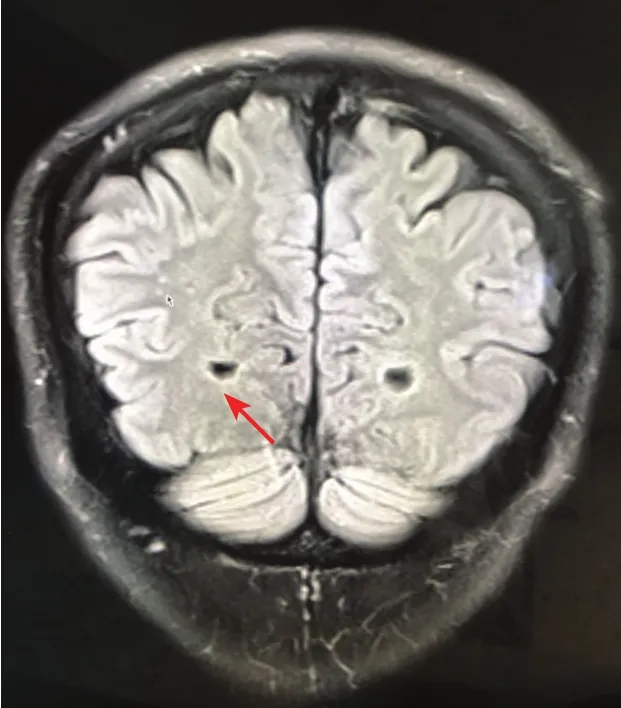
图3 法布里病患者颅脑磁共振成像
讨论
法布里病作为一种罕见的X连锁遗传溶酶体贮积症,其确切的患病率目前尚不清楚,普通人群中预估患病率为1/10万。因位于Xq22.1的GLA基因突变,导致其编码的α半乳糖苷酶A(α-galactosidase A,α-Gal A)活性降低或完全缺乏,造成代谢底物三己糖酰基鞘脂醇(globotriaosylceramides,GL-3)及其衍生物脱乙酰基GL-3(globotriaosylsphingosine,Lyso-GL-3)在肾脏、心脏、神经、皮肤等大量贮积,沉着于细胞胞质溶酶体内,特别是血管内皮细胞及周围细胞中级溶酶体内,引起相应的多脏器小血管病变。法布里病患者在儿童时期就会出现持续数分钟到数天的肢端神经痛,通常发生于运动后体温升高、发热或温热环境。患者经常伴有出汗异常,最常见的是无汗或少汗。其他表现可有泛发的皮肤血管角皮瘤、生长阻滞和青春期延迟,累及心血管系统可出现高血压、左心室肥厚、左室间隔增厚、心绞痛、心肌梗死、心瓣膜病、充血性心力衰竭等;累及消化系统可有腹痛,发作性腹泻、恶心、呕吐、里急后重;累及神经系统可出现短暂性脑缺血发作、中风、癫痫等;病情严重者可由于累及泌尿系统形成终末期肾病,导致过早死亡。患者机体所有组织细胞内均可见鞘糖脂沉积,血浆和尿沉淀出现酰基鞘鞍醇三己糖(GB3)和鞘氨醇球蛋白(globotriaosylsphingosine,lyso-GB3)升高[2]。法布里病的诊断需结合临床表现、酶活性、基因检测、生物标志物等多项指标。本例患者病程长达20余年,临床症状明显,基因检测及酶活性检测均有明确的阳性结果,是典型的法布里病。
目前临床上针对法布里病,主要有两种治疗方法:酶替代疗法和分子伴侣疗法[3]。酶替代疗法通过定期静脉注射人工重组a-GalA进行治疗,该酶可在细胞外通过和6-磷酸-甘露糖受体结合,有效水解溶酶体内沉积的GB3。重组人a-GalA的药物现有两种:agalsidase-α和agalsidase-β。这两种制剂均具有相似的糖基化模式、特异性活性和酶动力学,且均已被证明具有临床疗效[4]。但由于价格高昂,其在临床中的使用受到限制。分子伴侣疗法是通过一个小分子伴侣与突变酶蛋白结合,稳定蛋白构象或促进酶蛋白多肽的正确折叠。分子伴侣药物migalastat对GLA基因突变患者具有临床疗效,且最近已批准在美国使用[5]。其它治疗策略,如底物减少疗法、底物毒性干扰疗法和基因疗法,也正在研发中。对本例患者笔者主要采取定期复查,对症治疗等方法,目前患者各项指标稳定,病情未见明显进展。
猜你喜欢
中草药(2022年20期)2022-11-15
食品科学(2022年20期)2022-10-31
现代食品科技(2022年9期)2022-10-09
中国典型病例大全(2022年7期)2022-04-22
上海人大月刊(2022年4期)2022-04-14
医学前沿(2021年6期)2021-09-10
家庭科学·新健康(2020年6期)2020-07-06
中国美容医学(2019年2期)2019-03-13
丝路视野(2018年15期)2018-05-14
中国民族民间医药·下半月(2014年2期)2014-09-26
