
COL4A5基因新发突变致ALPORT综合征1例并文献复习
2022-05-10 07:49孙婉林毅张冉冉邵乐平张秋业
精准医学杂志 2022年1期
孙婉 林毅 张冉冉 邵乐平 张秋业
(1 青岛大学附属医院儿科,山东 青岛 266003; 2 青岛市市立医院肾脏科)
ALPORT综合征(ALPORT syndrome, AS)是由编码肾小球基底膜Ⅳ型胶原蛋白的基因突变导致的遗传性肾脏疾病[1]。肾小球基底膜Ⅳ型胶原α链的α3、α4以及α5链分别由COL4A3、COL4A4以及COL4A5基因编码[2],上述基因突变可导致患者出现血尿、蛋白尿及慢性进行性肾功能减退,部分患者可以表现为高频神经性耳聋、近视等肾外表现[3-4]。由于缺乏有效治疗手段,该病预后较差,最终多进展为终末期肾病(ESRD)[5]。现有研究表明,ESRD发生的早晚及进展速度取决于治疗开始时疾病所处的阶段或状态[6]。因此,AS的早期诊断与合理干预非常重要。本研究报道了1例由COL4A5基因新发突变导致的AS,并对患者的分子遗传学资料进行了分析,以期提高对本病的认识,为本病的诊断和临床干预提供参考。
1 临床资料
患者,男,8岁,因“尿检异常6月余”于2019年11月3日入住我院心肾免疫儿科。患者2019年4月18日于“上呼吸道感染”后1周出现肉眼血尿伴轻微眼睑浮肿,就诊当地儿童医院。尿常规显示隐血,红细胞10.21×109/L,蛋白,血清补体C3 0.40 g/L,抗链球菌溶血素O(ASO)316 kU/L,血清免疫球蛋白IgG 5.59 g/L,24 h尿蛋白定量为53 mg/kg,尿红细胞形态检查显示严重变形红细胞占40%。疑诊为“急性链球菌感染后肾小球肾炎”,采用青霉素类药物抗感染及对症治疗2周,尿蛋白及血尿情况未见好转。随后给予甲泼尼龙[10 mg/(kg·d),连用3 d]治疗2个周期,治疗后患者肉眼血尿消失,予以出院。出院后每天1次口服福辛普利(10 mg);继续口服强的松[1 mg/(kg·d)]治疗,后逐渐减量,于2019年10月15日停用。2019年8月复查血清补体C3、ASO及血清IgG均恢复正常,尿常规显示尿蛋白、尿红细胞10.20×107/L,偶有晨起眼睑浮肿,尿量基本正常。为进一步诊治,2019年11月3日就诊于我院门诊,以“肾炎综合征”收入院。
入院体格检查:体温36.5 ℃,脉搏92 min-1,呼吸23 min-1,体质量38 kg,血压122/63 mmHg。神志清,精神好,全身无皮疹,无颜面及眼睑水肿,肾区无叩击痛,双下肢无浮肿。余体格检查未见明显异常。辅助检查:尿液分析显示尿蛋白,隐血,红细胞10.20×107/L;24 h尿蛋白定量测定为35.7 mg/kg;血生化检查结果示总蛋白50.31 g/L,白蛋白32.61 g/L,球蛋白17.7 g/L,谷丙转氨酶23 U/L,谷草转氨酶30 U/L,尿素氮3.84 mmol/L,肌酐30 μmol/L,总胆固醇3.96 mmol/L,低密度脂蛋白2.19 mmol/L,三酰甘油1.21 mmol/L;血清IgG 4.09 g/L,IgA 1.02 g/L,IgM 0.79 g/L,IgE 14.39 kU/L;血清补体C3 1.29 g/L,C4 0.28 g/L;血常规、血清电解质、调节性淋巴细胞亚群、红细胞沉降率、血凝指标等检测无异常。听力传导通路检查未见异常。肾脏组织穿刺活检病理学检查结果:①光镜下苏木精-伊红(HE)染色结果显示,毛细血管袢开放良好,节段系膜区轻度增宽,系膜细胞与基质增多,囊壁增厚(图1A);过碘酸六胺银染色(PASM)、Masson染色均阴性;糖原染色(PAS)显示肾小管间质病变轻,小灶状小管上皮细胞扁平、刷状缘脱落,偶见蛋白管型,肾间质较多泡沫细胞聚集(图1B);肾脏组织免疫荧光染色检查(未进行Ⅳ型胶原α链免疫荧光染色检查):IgG、IgA、IgM、C3、C1q、λ链、κ链、PLA2R均阴性;病理诊断:肾小球节段系膜轻度增生性病变;②普通透射电子显微镜检查:肾小球基底膜厚薄不一,基底膜致密层增厚,部分呈撕裂状和蛛网状,足突部分融合,未见电子致密物沉积(图1C、D)。
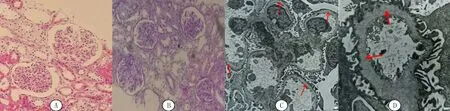
A:光镜下肾脏组织HE染色结果,200倍;B:光镜下肾脏组织PAS染色结果,100倍;C:普通透射电子显微镜照片,基底膜致密层分层状改变,5 000倍;D:普通透射电子显微镜照片,毛细血管袢明显撕裂分层,呈蛛网状,12 000倍晶状体和斑点状视网膜病变,前者临床表现为进行性加重的近视,后者一般不会影响视力,多不需要进行治疗[12]。
为进一步明确病因,经患者家属知情同意后,采集患者及其父亲和母亲外周血各2 mL,提取DNA,靶向探针捕获,选择增强全外显子组点突变项目对患者进行高通量二代测序,患者父亲和母亲均采用一代测序技术(Sanger)对同一可疑的致病变异进行验证(由深圳安吉康尔医学检验实验室协助完成)。患者基因检测结果显示X染色体长臂2区2带3亚带(Xq22.3)上的COL4A5基因存在一处半合子变异(c.3393_3419del),即第38号外显子编码区第3393~3419位碱基缺失,导致第1128~1136号氨基酸缺失(p.G1128_S1136del)(图2A),该突变为框内缺失突变,患者父母该位点均无变异(图2B、C)。在OMIM、Pubmed、HGMDpro、Clinvar、dbSNP、Varcards等数据库对该突变位点进行检索,均未见收录,为首次发现的新发突变。根据美国医学遗传学与基因组学会(ACMG)发布的基因序列变异解读指南[7]进行致病性分析,同时结合患者临床表现及典型肾脏组织病理学特征,判定该突变为致病性突变。
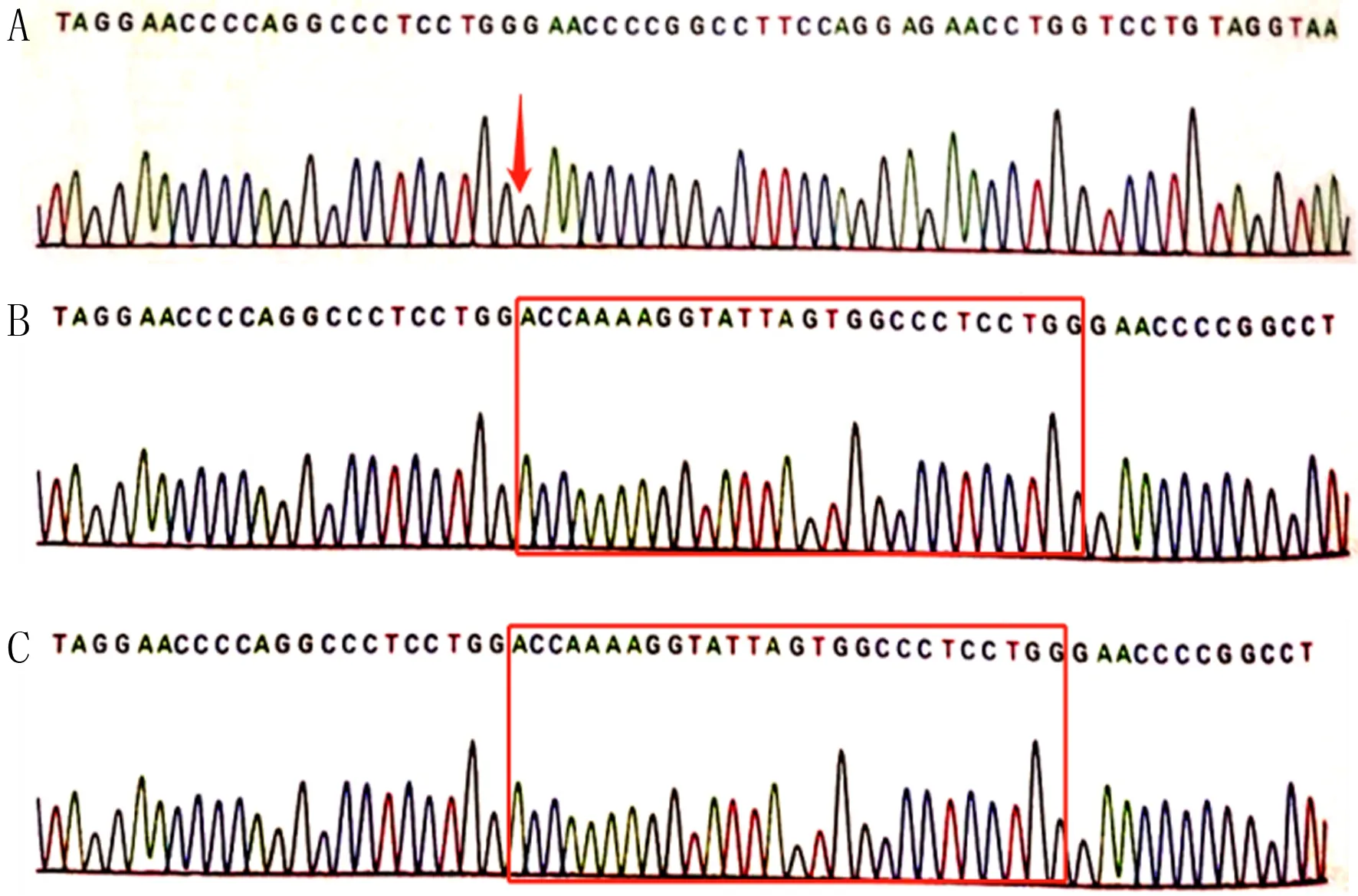
A:患者,箭头处27个碱基缺失(c.3393_3419del);B:患者父亲;C:患者母亲
根据患儿病史、体征、相关辅助检查、肾脏组织病理及基因检测结果,明确诊断为AS。继续每天一次口服福辛普利(10 mg),以减少尿蛋白分泌、延缓肾脏病变,门诊随访半年。随访期间患儿尿常规显示尿蛋白,隐血,红细胞8.28×108/L,肾功能尚未受损。眼科及耳鼻咽喉科门诊随访过程中无感音神经性耳聋,无近视、斜视、圆锥形角膜、眼底病变等情况出现。
2 讨 论
AS是儿童最常见的遗传相关性肾炎[8]。多在10岁前以肾小球源性血尿起病[9],起病隐匿,随着时间进展可伴有不同程度蛋白尿,肾功能呈进行性衰竭。除了严重的肾脏损害,AS也可引起听力损伤及眼部损伤。其中听力损伤的发生率为50%~67%[10],可为AS患者的首发症状之一,且多为双侧对称性感音性耳聋,最初多累及高频区,而后进行性累及全音域[11]。AS的眼部损伤主要包括圆锥状AS的诊断标准为持续性肾小球性血尿或血尿伴蛋白尿的患者符合以下任意一条标准即可确诊:电镜下肾小球基底膜致密层撕裂分层、肾小球基底膜Ⅳ型胶原α3、α4、α5链免疫荧光染色异常、皮肤基底膜Ⅳ型胶原α5链免疫荧光染色异常、基因检测发现COL4A5基因有一个致病性突变或COL4A3/4基因有两个致病性突变[5]。但本研究认为肾组织电镜检查及Ⅳ型胶原α链免疫荧光染色检查均为间接性结果,疾病早期症状不典型,易造成漏诊。基因检测不仅有助于明确突变类型、确诊疾病,也可为该病的产前诊断提供线索[13-14]。本研究中患者为8岁男孩,慢性病程,以肉眼血尿及大量蛋白尿起病,糖皮质激素治疗无效,结合肾脏活检组织电镜观察,显示肾小球基底膜厚薄不一,基底膜致密层增厚,部分呈撕裂状和蛛网状;基因检测发现COL4A5基因存在致病性缺失突变,可明确AS诊断。该患者总病程6个月,病初发现补体C3下降,ASO轻微升高,后当地医院门诊随诊时完善补体及ASO检查,发现指标恢复正常,因此该患者病初不能排除合并急性链球菌感染后肾小球肾炎的可能性。
AS远期预后不佳,男性尤为突出,常在20~30岁进展至ESRD[15],进行性加重的蛋白尿为导致AS不良预后的主要相关因素[16]。也有学者发现,AS患者肾脏损伤程度与听力受损程度具有正相关性[17]。该患者以肾脏损伤为主,且程度较重,因此住院期间于我院耳鼻咽喉科行畸变产物耳声发射检查,显示听力传导通路未见异常。推测一方面可能与其年龄尚幼、临床表型尚不完全有关。另一方面,感音神经性耳聋起病较为隐匿,临床多采用纯音测听、声导抗、畸变产物耳声发射、听性脑干反应等方法联合进行早期筛查,而该患者仅完善了畸变产物耳声发射检查。该检查是一种评价外周听觉通路的方法,可精确地反映耳蜗毛细胞在相应频率上的功能状态;但检测灵敏度较低,可能存在漏诊的可能。
根据遗传方式的不同,AS可分为以下3种类型[18],COL4A5基因突变所致的X连锁显性遗传性AS(XLAS)、COL4A3或COL4A4基因突变所致的常染色体隐性遗传AS(ARAS)及常染色体显性遗传AS(ADAS),其中以XLAS最常见,大约占85%。检索HGMD数据库中COL4A5基因突变的患者,迄今为止,有1 286个COL4A5基因突变位点,尚未见有热点突变。其中错义/无义突变620个(错义突变占41.8%左右,无义突变占6.3%左右),缺失突变为342个(26.6%),剪接突变为222个(17.3%),插入突变74个(5.8%),插入/缺失突变13个(1.0%),插入/重复突变7个(0.5%),复杂重组8个(0.6%)。缺失或插入导致移码突变占7.8%左右。本研究中患者基因检测显示COL4A5基因第38号外显子编码区第3393~3419位碱基缺失,导致第1128~1136号氨基酸缺失,并致蛋白质结构发生改变,之后16个氨基酸后出现终止密码子,该突变类型为整码突变。根据ACMG指南[7]分析:该突变为新发突变,经家系验证,其父母在该位点均无变异,为强致病性证据(PS2);该突变为非重复区域中的阅读框内插入/缺失或终止密码子丧失导致蛋白质长度改变,为中等致病性证据(PM4);该突变在gnomAD数据库、千人数据库、ExAC数据库的正常人群中未发现(或极低频位点),是罕见变异,为中等致病性证据(PM2)。联合上述证据,确定其变异性质为PS2+PM2+PM4,初步判定为“可能致病性变异”中的组合b。结合患者已出现持续性血尿、蛋白尿等临床表现及典型肾脏组织病理学特征,提示该突变已导致蛋白质结构及功能改变,因此该突变为致病性突变。该基因突变遗传方式为X连锁显性遗传,基因突变信息应该仅遗传给其女儿。
目前相关研究表明,COL4A5基因突变的类型、位置不同,则相应临床表型、ESRD进展速度也不同[18-24]。无义突变、移码突变及供体剪接突变等突变类型的患者相应临床表型较为严重,20岁左右即可进展为ESRD,且大多合并肾外损害,其中听力异常可达80%。第21~47号外显子甘氨酸突变、非甘氨酸错义突变、受体剪接突变及不引起移码突变的缺失或插入病变患者临床表型严重程度较前者次之,多在25岁左右进展为ESRD,其肾外损害较少。而发生在第1~20号外显子甘氨酸替代突变的患者临床症状往往较轻,ESRD患者多于30岁之后发生[18,24]。本研究中患者COL4A5基因第38号外显子编码区存在致病性缺失突变,在幼年期即存在持续血尿伴间断大量蛋白尿,无听力、眼部损害等肾外表现,与上述研究相符;该患者目前长期口服福辛普利治疗,尚未出现肾衰竭症状,后续需密切随访。
综上所述,临床上不明原因的血尿、蛋白尿患者,若糖皮质激素治疗无效,或存在肾功能慢性进行性损伤、听力受损、眼部异常等肾外表现,建议尽早行肾脏穿刺活检和(或)基因检测以明确病因。AS的早期诊断有助于避免使用糖皮质激素及免疫抑制剂带来的副作用,可以更加合理地选择治疗方案及评估患者预后。另外,对于患者家属的再生育也具有一定的指导作用。
利益冲突声明:所有作者声明不存在利益冲突。
ConflictsofInterest: All authors disclose no relevant conflicts of interest.
伦理批准和知情同意:本研究涉及的所有试验均已通过青岛大学附属医院医学伦理委员会的审核批准(文件号QYFYWZLL26836)。所有试验过程均遵照《人体医学研究的伦理准则》的条例进行。受试对象或其亲属已经签署知情同意书。
EthicsApprovalandPatientConsent: All experimental protocols in this study were reviewed and approved by The Medical Ethics Committee of The Affiliated Hospital of Qingdao University (Approval Letter No.QYFYWZLL26836), and all experimental protocols were carried out by following The Ethical Guidelines for Human Medical Research. Consent letters have been signed by the research participants or their relatives.
作者贡献:张秋业、孙婉、张冉冉、邵乐平参与了研究设计;孙婉、林毅、张秋业参与了论文的写作和修改。所有作者均阅读并同意发表该论文。
Contributions: The study was designed byZHANGQiuye,SUNWan,ZHANGRanran, andSHAOLeping. The manuscript was drafted and revised bySUNWan,LINYi, andZHANGQiuye. All the authors have read the last version of the paper and consented submission.
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
听力学及言语疾病杂志(2022年4期)2022-07-15
祝您健康·文摘版(2021年12期)2021-12-08
中国土壤与肥料(2021年5期)2021-12-02
中华耳科学杂志(2021年1期)2021-02-01
实用癌症杂志(2020年3期)2020-03-19
复旦学报(医学版)(2016年1期)2016-06-20
中国动物保健(2015年4期)2015-10-21
祝您健康(2000年11期)2000-12-31
祝您健康(1987年6期)1987-12-31
