
玉竹多糖低共熔溶剂提取工艺优化及其抗氧化和抗糖基化活性研究
2022-05-07 13:51何瑞阳王锋苏小军李清明杨华朱晓慧范宽秀江雪梅
食品与发酵工业 2022年8期
何瑞阳,王锋*,苏小军,李清明,杨华,朱晓慧,范宽秀,江雪梅
1(湖南农业大学 食品科学技术学院,湖南 长沙,410128)2(湖南省发酵食品工程技术研究中心,湖南 长沙,410128) 3(湖南农业大学 生物科学技术学院,湖南 长沙,410128)
玉竹(Polygonatumodoratum)为百合科黄精属玉竹的根茎,又名葳蕤、铃铛菜、尾参,是我国重要的药食同源大宗药材,具有滋阴润燥、生津止咳的功效。现代药理学研究表明,玉竹具有抗氧化、调节免疫力、降糖、降血压等作用[1]。玉竹含有丰富的多糖、黄酮、多酚等成分[2],其中,多糖是其主要活性成分,在医药、食品、化妆品行业具有广阔的应用前景[3]。
低共熔溶剂(deep eutectic solvents, DESs)自ABBOTT等[4]2003年首次提出之后,由于其具有成本低、易合成、高溶解度、微毒甚至无毒等优点[5],在生物活性成分提取领域受到广泛关注。其中,由氯化胆碱作为氢键受体(hydrogen-bond acceptor,HBA)与其他氢键供体(hydrogen-bond donor,HBD)组成的DESs在黄酮、多酚等生物活性物质的提取方面[3]研究较多。也有文献报道用于植物活性多糖的提取方面,如梁静[6]采用氯化胆碱与甘油组合DESs提取铁皮石斛多糖,提取率高达44.35%,显著高于传统提取方式。唐兰芳[7]采用氯化胆碱与1,4-丁二醇组合提取黄精多糖(Polygonatumsibiricunmpolysaccharide extracted by deep eutectic solvents, DPsP),最终提取率可达33.81%,约为热水浸提法的4倍。DESs自身的极性、黏度等对多糖提取率起决定性作用;除此之外,提取过程中的温度、时间与液料比等也对多糖的提取率有影响[8],所以在利用DESs提取植物活性多糖时,对提取条件进行优化以获得最优提取条件至关重要。
大多数关于玉竹的研究,均是热水提取[1],鲜有采用DESs提取玉竹多糖(Polygonatumodoratumpolysaccharide extracted by deep eutectic solvents, DPoP)的研究报道。本研究采用DESs提取玉竹中的活性多糖,比较不同DESs组合,研究DESs摩尔比、DESs含水量、提取温度、提取时间、液料比等因素对多糖提取率的影响,通过响应面优化工艺参数,采用DPPH法、ABTS法、氧化自由基吸收能力(oxygen radical absorbance capacity,ORAC)法对其进行抗氧化活性测定,并测定DPoP对糖基化终末产物(advanced glycation end products,AGEs)生成的抑制作用,旨在建立一种玉竹多糖的高效提取方法,并为玉竹多糖的开发利用提供理论依据和技术参数。
1 材料与方法
1.1 材料与试剂
玉竹(品种:猪屎尾),产自湖南郴州桂阳;葡萄糖(纯度≥99%),上海麦克林生化科技有限公司;DPPH(纯度>97%),梯希爱(上海)化成工业发展有限公司;ABTS(纯度≥98%),上海瑞永生物科技有限公司;2,4,6-三吡啶基三嗪(2,4,6-tripyridin-2-yl-1,3,5-triazine,TPTZ,纯度≥98%),上海瑞永生物科技有限公司;氨基胍(纯度≥98%),美国Sigma公司;乙醇(含量≥99.7%),成都市科隆化学品有限公司;考马斯亮蓝G-250、牛血清蛋白(bovine serum albumin,BSA)、无水葡糖糖、氯化胆碱、尿素、硫酸、甲醇等均为分析纯,国药集团化学试剂有限公司。
1.2 仪器与设备
AL204电子天平,梅特勒-托利多仪器(上海)有限公司;BJ-100型高速多功能粉碎机,德清拜杰电器有限公司;101A-3ET电热鼓风干燥箱,上海实验仪器厂有限公司;SCIENTZ-18 N冷冻干燥机,宁波新芝生物科技股份有限公司;TGL16M冷冻离心机,长沙英泰仪器有限公司;RE-2000B旋转蒸发器,巩义市予华有限责任公司;SHZ-D(I)W循环水式多用真空泵,上海力辰邦西仪器科技有限公司;UV1901G/UV1901PCS紫外可见分光光度计,上海佑科仪器仪表有限公司;H-8数显恒温水浴锅,上海浦东物理光学仪器厂;VarioskanFlash多功能读数仪,美国赛默飞世尔科技有限公司;IRAffinity-1傅立叶红外光谱仪,日本岛津有限公司;F-7000荧光分光光度计,日本日立公司。
1.3 实验方法
1.3.1 原料预处理
新鲜玉竹洗净、去须、切片,放入烘箱中,60 ℃烘干至水分含量10%以下,使用高速多功能粉碎机粉碎,过70目筛得玉竹粗粉,低温密封保存备用。
1.3.2 玉竹多糖提取
(1)热水浸提法(Polygonatumodoratumpolysaccharide extracted by hot water,HW):根据《国家药典》第三部(2015版),称取1 g玉竹粗粉置于圆底烧瓶内,加入蒸馏水100 mL,加热回流1 h,脱脂棉过滤,重复提取2次,滤液合并浓缩置100 mL容量瓶定容,得水提多糖溶液(HWPoP)。
(2)DESs提取法:称取1 g玉竹粗粉置于三角瓶中,以一定比例加入DESs,热水浴提取,提取结束冷却后加入4倍体积无水乙醇,4 ℃醇沉12 h后离心、复溶于100 mL蒸馏水,得DESs提取多糖溶液(DPoP)[9]。
1.3.3 玉竹多糖提取率计算
葡萄糖标准曲线制备:采用苯酚-硫酸法制作标准曲线,以吸光度为纵坐标,浓度为横坐标,绘制标准曲线。回归方程为y=26.337x-0.003 9,R2=0.999 5。
按照1.3.2提取操作各重复3组,采用苯酚-硫酸法比对标准曲线测定多糖含量,按照公式(1)计算多糖提取率:
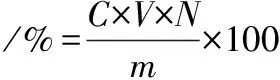
(1)
式中:C,通过标准曲线计算得多糖质量浓度,mg/mL;V,样品体积,mL;N,稀释倍数;m,称取玉竹粗粉质量,g。
1.3.4 不同组合DESs对玉竹多糖提取率的影响
以多糖提取率为指标,选取酰胺(尿素)、羧酸(丁二酸、柠檬酸)和多元醇(1,4-丁二醇、乙二醇)作HBD,氯化胆碱作HBA,以摩尔比2∶1的比例配成5种不同组合的均匀稳定的DESs溶液[10],在温度90 ℃、液料比30∶1、提取时间40 min,按照1.3.2(2)提取方法提取玉竹多糖,并计算提取率。5种DESs组合见表1。

表1 DESs类型Table 1 Type of DESs
1.3.5 单因素实验设计
从1.3.4提取结果中选择提取率最高的DESs组合,在本实验室的研究基础上选取DESs摩尔比(1∶1、2∶1、3∶1、4∶1、5∶1、6∶1)、提取温度(50、60、70、80、90、100 ℃)、提取时间(20、30、40、50、60、70 min)、溶剂含水量(0%、10%、20%、30%、40%、50%)、液料比(10∶1、20∶1、30∶1、40∶1、50∶1、60∶1)进行单因素考察,考察各因素对提取率的影响。
1.3.6 响应面优化实验设计
在单因素考察结果上固定DESs摩尔比,对提取温度(A)、提取时间(B)、溶剂含水量(C)、液料比(D)4个单因素进行响应面优化实验,以多糖提取率作响应值进行响应面Box-Behnken(BBD)分析,确定多糖提取最优条件组合。响应面实验因素与水平设计见表2。

表2 响应面实验因素与水平设计Table 2 Factors and level of Box-Behnken
1.3.7 DPoP制备
参照宗鑫妍等[1]的方法,采用Sevage试剂对1.3.2制备的DPoP溶液进行脱蛋白处理,并采用考马斯亮蓝法测定处理后溶液中可溶性蛋白含量。以BSA作对照,在吸光度595 nm处,以吸光度为纵坐标,蛋白浓度为横坐标,绘制可溶性蛋白标准曲线,回归方程为y=5.162 9x+0.040 5,R2=0.999 2。重复脱蛋白操作直至上层多糖溶液中可溶性蛋白含量稳定。经脱蛋白处理的DPoP溶液浓缩至一定体积后加入4倍体积无水乙醇,4 ℃冷藏6 h后离心、冻干得DPoP粉末,将其低温贮藏备用。
1.3.8 DPoP红外光谱测定
取1 mg低温烘干的DPoP粉末,加入100 mg烘干至恒重的KBr,置研钵中研磨均匀,压片,在波数4 000~400 cm-1内进行红外扫描。
1.3.9 玉竹多糖抗氧化活性测定
DPPH自由基清除能力测定参照ZHAO等[11]的方法进行;ABTS阳离子自由基清除能力测定参照CHENG等[12]的方法进行;ORAC法参照ZAR等[13]的方法进行。
1.3.10 DPoP抗糖基化能力测定
参照张家成[14]的方法稍做改动,测定多糖的抗糖基化能力,配制20 mg/mL BSA溶液、0.5 mol/L葡萄糖溶液(glucose,Glu),按照体积比1∶1混合过无菌滤膜,取3 mL BSA-Glu混合液,分别加入样品1 mL与6.0 mL 0.2 mol/L磷酸缓冲溶液,摇匀后避光37 ℃ 恒温孵育6 d,以氨基胍代替样品做对照组,磷酸缓冲溶液代替样品做空白对照,测定AGEs在激发波长370 nm,发射波长440 nm处的荧光值,玉竹多糖对AGEs的相对抑制率按公式(2)计算:

(2)
式中:Fc,空白组荧光值;Fs,样品组荧光值;Fs1,反应体系由缓冲溶液代替Glu样品的荧光值;Fs2,反应体系由缓冲溶液代替BSA样品的荧光值。
1.4 数据分析
所有操作均重复3次,实验数据采用“平均值±标准偏差”表示,采用SPSS 19 LSD法进行方差分析,P<0.05为差异显著,采用Origin 2018进行绘图。
2 结果与分析
2.1 不同组合DESs对多糖提取率的影响
如图1所示,多糖提取率依次是UCC>EGCC>BCC>HW>AACC>CACC,尿素与氯化胆碱组合DESs对多糖的提取率显著高于其他组合,氯化胆碱与多元醇组合DESs提取率高于HW,氯化胆碱与羧酸组合DESs则低于HW,这与汪涛等[15]在提取DPsP时结果类似,引起提取效果差异的原因可能是不同DESs之间的扩散度、溶解度、黏度、极性等理化性质不同。实验确定以尿素与氯化胆碱组合做后续优化。
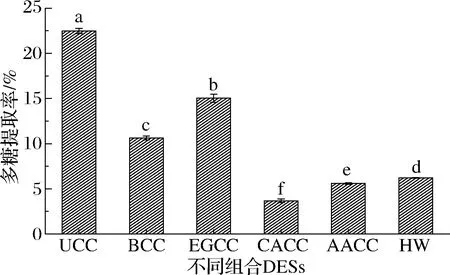
图1 不同组合DESs对多糖提取率的影响Fig.1 Effects of different types of DESs on the yield of polysaccharide 注:不同字母代表差异显著(P<0.05),相同字母代表差异不显著
2.2 单因素试验结果
2.2.1 尿素与氯化胆碱摩尔比对提取率的影响
如图2所示,当尿素与氯化胆碱摩尔比开始增大时,多糖提取率呈现增长趋势,在3∶1时达到最大,超过3∶1之后,多糖提取率降低,这与尿素中的NH2基团与Cl-形成的氢键强弱对DESs的熔点与黏度造成的影响有关[16],在摩尔比为5∶1时的提取率出现上升现象,而夏伯候等[17]在采用1,2-丙二醇与氯化胆碱组合提取夏枯草总黄酮时于摩尔比5∶1处也出现类似现象,具体原因有待进一步研究。本研究选择尿素与氯化胆碱摩尔比3∶1做后续优化。
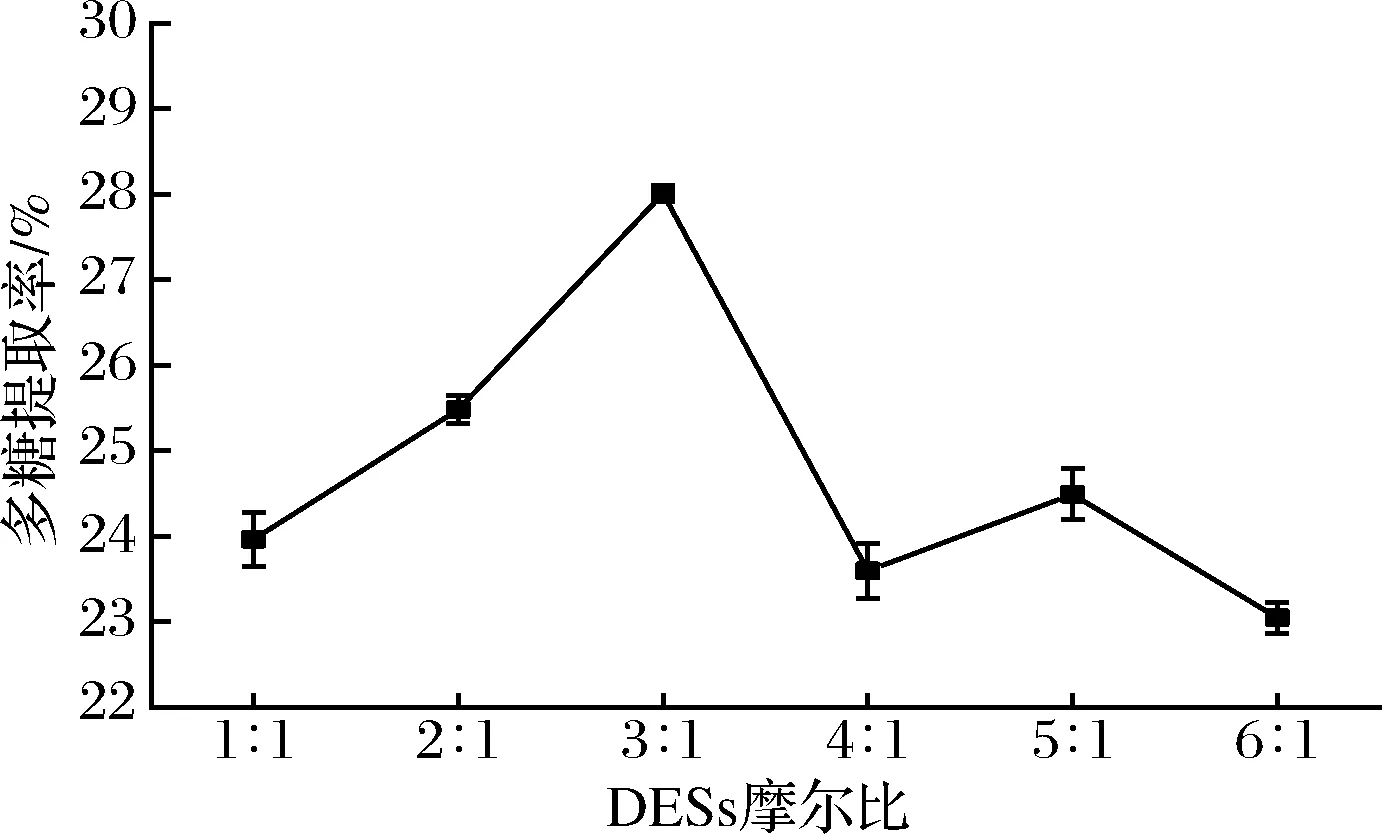
图2 DESs摩尔比对多糖提取率的影响Fig.2 Effects of DESs molar ratio on the yield of polysaccharide
2.2.2 提取温度对提取率的影响
如图3所示,随着温度的升高,多糖提取率不断增大,当温度超过90 ℃以后多糖提取率呈现下降的趋势,可能是高温导致的多糖降解,温度对DESs的黏度、极性和物料的溶解度都会造成影响[18],所以选择提取温度90 ℃做后续优化。

图3 提取温度对多糖提取率的影响Fig.3 Effects of temperature on the yield of polysaccharide
2.2.3 提取时间对提取率的影响
如图4所示,随着提取时间延长提取率快速上升,在40 min时达到最大,提取时间超过40 min后提取率降低,在一定时间内,提取时间与活性成分溶出率成正比,超过40 min后可能出现多糖结构破坏降解,导致提取率降低,而在60 min时较50 min时的提取率略有升高,但不具有显著性(P>0.05),可能是多糖继续溶出与结构破坏共同作用的结果。本研究选择提取时间40 min做后续优化。
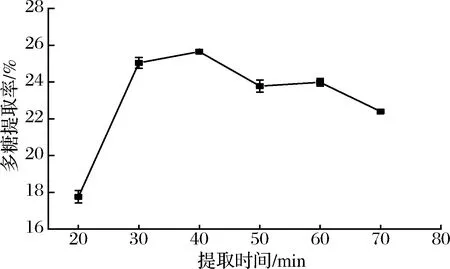
图4 提取时间对多糖提取率的影响Fig.4 Effects of time the yield of polysaccharide
2.2.4 DESs含水量对提取率的影响
如图5所示,当DESs水分含量在0%~20%时,提取率随含水量增大而增大,在20%时达到最大,当含水量超过20%,提取率开始下降。含水量在30%~40%出现上升趋势,然而有趣的是宋歆睿[19]在采用低共熔溶剂提取蒲公英主要活性成分时,目标物的提取率在含水量为40%时较含水量30%时也有所上升,这可能是由于未结合的水降低了DESs的黏度与表面张力,提取率出现上升现象,而含水量继续增加导致分子间作用力降低,提取率迅速下降[20]。本研究选择DESs含水量20%做后续优化。

图5 DESs含水量对多糖提取率的影响Fig.5 Effects of DESs water content the yield of polysaccharide
2.2.5 液料比对提取率的影响
如图6所示,液料比在10∶1至30∶1时,溶剂量的增加,使得物料细胞与外界溶剂之间造成浓度差,浓度差增加更易使得植物细胞破碎,多糖提取率呈上升趋势,而当液料比超过30∶1时,过多的DESs可能使得物料之间相互作用减弱,不利于多糖提取,所以选择提取液料比30∶1做后续优化。
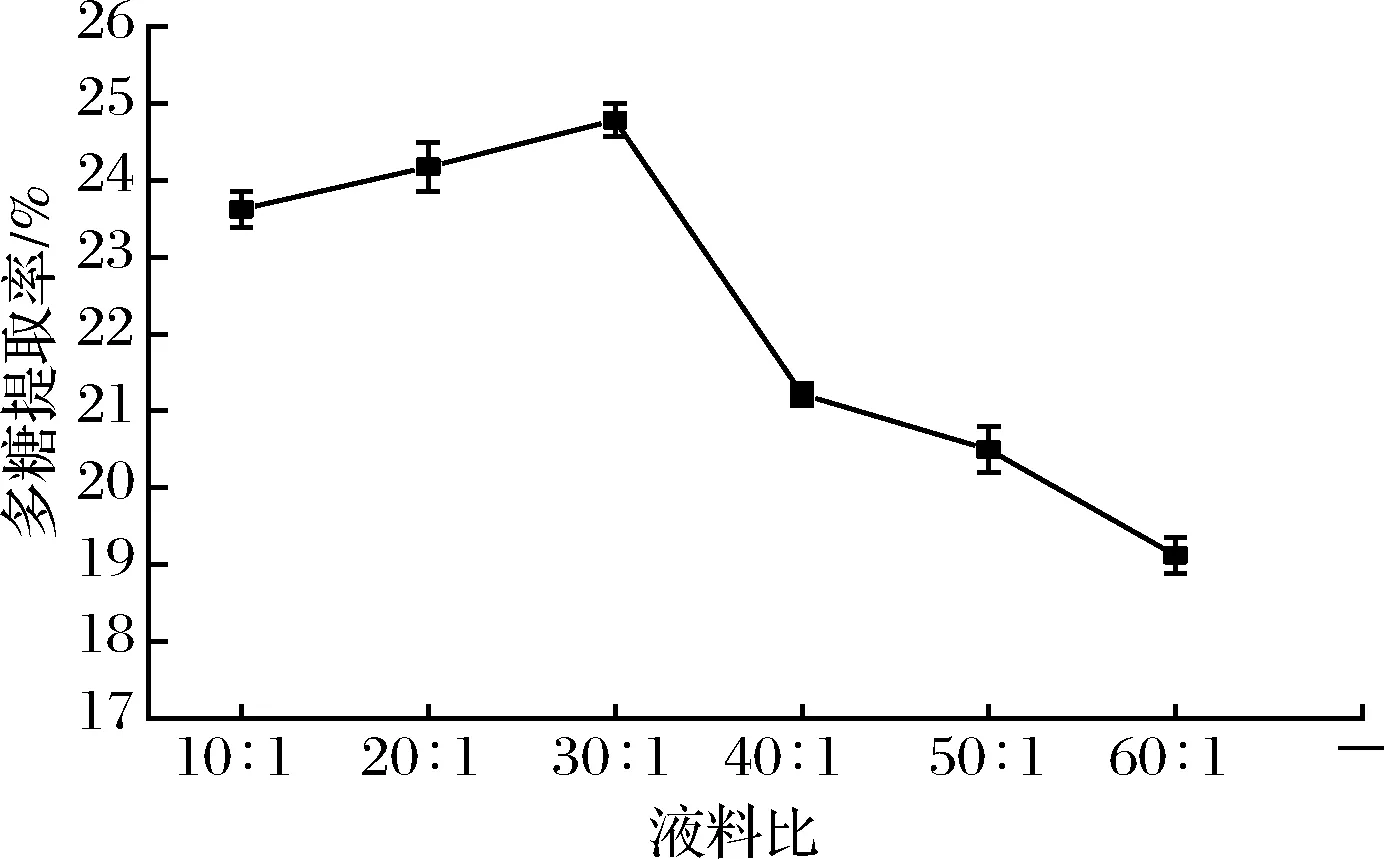
图6 液料比对多糖提取率的影响Fig.6 Effects of liquid-solid ratio on the yield of polysaccharide
2.3 响应面试验结果分析
根据2.2结果分析,固定尿素与氯化胆碱摩尔比为3∶1,选取提取时间、提取温度、DESs含水量与液料比作为单因素,选取多糖提取率为响应值,进行响应面优化实验,Box-Behnken实验设计与结果见表3。
2.3.1 回归方程模型与方差分析
采用Design-Expert 8.0对数据进行回归拟合分析,二次方程模型为Y=29.69+0.76A+0.46B-0.35C-0.36D-0.14AB-0.58AC+0.54AD+0.14BC-0.29BD+1.02CD-2.20A2-2.30B2-3.09C2-3.07D2。
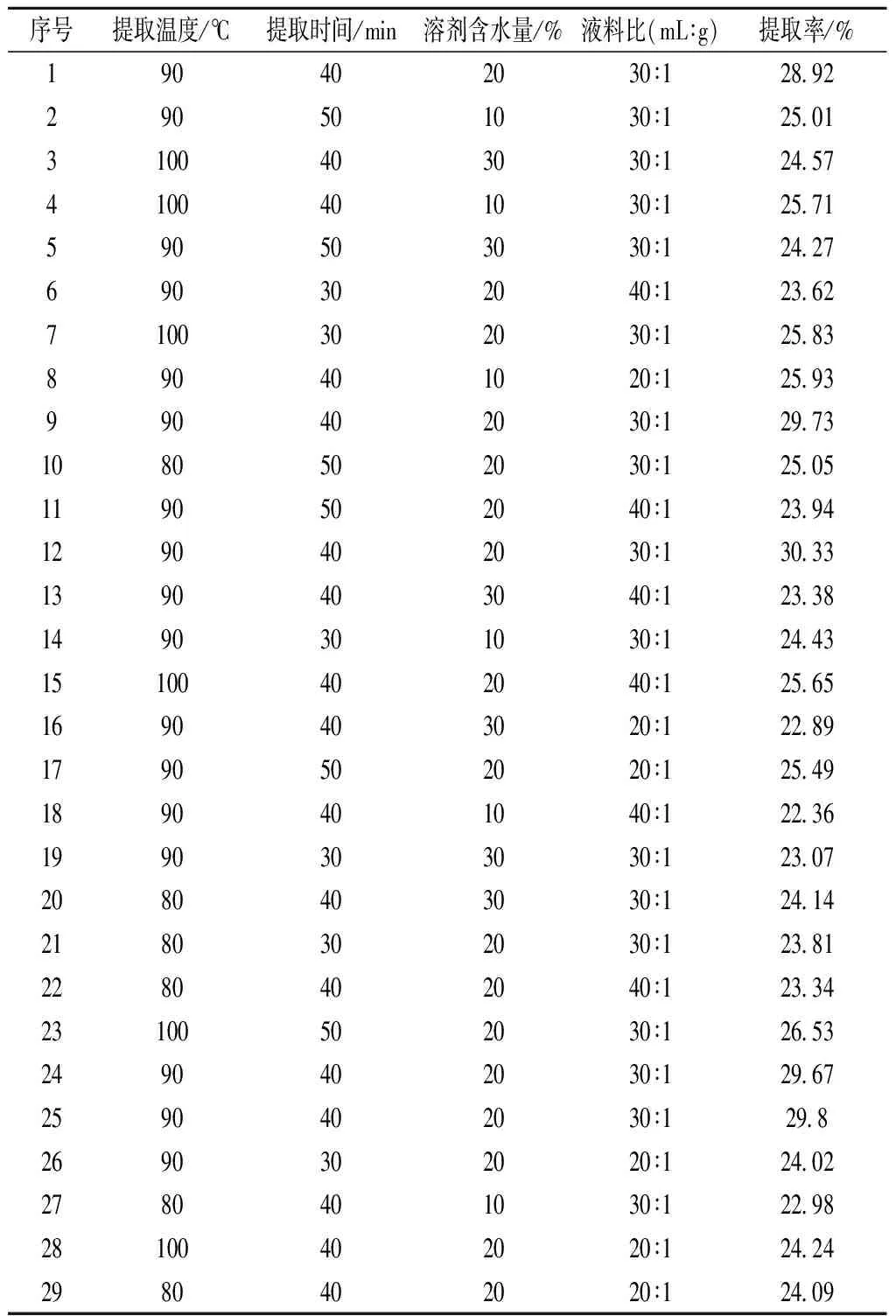
表3 响应面试验设计与结果Table 3 Design and result Box-Behnken
根据表4结果分析,回归模型总回归系数R2=0.973 4,且模型P<0.01,实验模型达到极显著水平,模型方差失拟项P=0.492 6>0.05,说明该模型拟合度良好,可用于预测4种因素不同组合情况下多糖的提取率,模型变量A、B、CD、A2、B2、C2、D2均达极显著水平(P<0.000 1),单因素变量C、D及相交变量AC均达显著水平(P<0.05),各单因素在取值范围内,对玉竹多糖提取率影响大小顺序为:提取温度(A)>提取时间(B)>液料比(D)>溶剂含水量(C)。
2.3.2 响应面分析
通过Design-ExPert8.0对数据进行拟合得到响应面图,响应面的三维图与等高线图可以看出2个变量之间的相互作用,如图7-a所示,随着提取温度与含水量的增加,提取率呈现先增大后减小的趋势,等高线图密集且呈椭圆形,表明两变量相交对提取率影响极显著,如图7-b所示,随着含水量与液料比的增大,提取率呈现先增大后减小的趋势,等高线图呈椭圆形,证明两变量相交对提取率影响显著,如图7-c所示,随着提取温度与液料比的增大,提取率也呈现先增后减的趋势,三维图呈凸面,等高线图椭圆形,但相比图7-a,等高线较为稀疏,证明两变量相交对提取率有一定影响,这与方差分析结果一致。
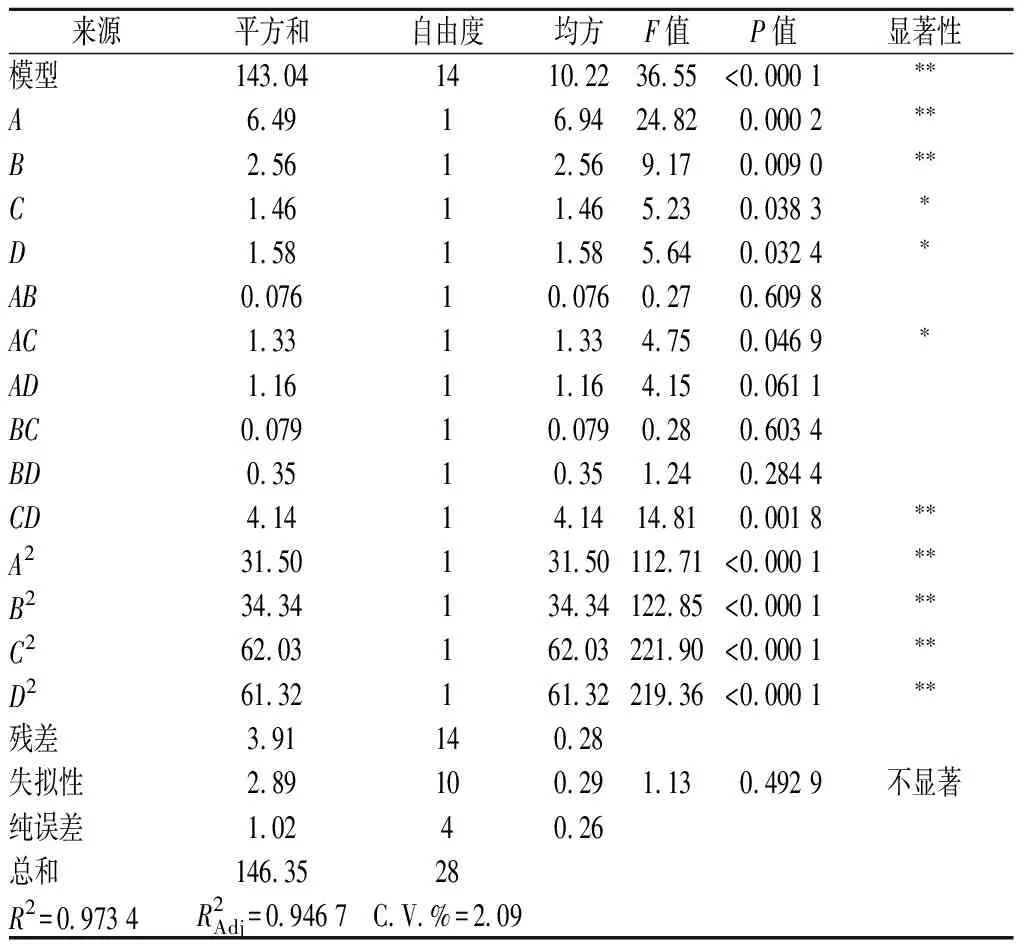
表4 响应面结果分析Table 4 Analysis of Box-Behnken
2.3.3 回归模型验证实验
通过响应面优化分析,得到DESs提取玉竹多糖预测最优工艺条件为:提取温度91.73 ℃、提取时间40.97 min、溶剂含水量19.2%,液料比29.38∶1,预测多糖提取率为29.80%,为验证回归模型可信性,采取上述最佳操作条件进行提取实验,考虑到实际操作与仪器限制,将最终选取提取条件设为:摩尔比3∶1、提取温度92 ℃、提取时间41 min、溶剂含水量19%,液料比29∶1,在此优化条件下,多糖提取率为(29.03±0.54)%,与预测值接近,表明该实验回归模型可靠,能较为准确的预测玉竹多糖提取率。多糖提取率为热水浸提法(6.21±0.05)%的4.67倍。
2.4 玉竹多糖红外光谱测定结果分析


图8 DPoP红外光谱图Fig.8 FTIR of spectra of DPoP
2.5 DPoP对DPPH自由基清除能力
自由基由在人体内积累会引发某些慢性疾病,如动脉粥样硬化、神经病变、慢性抑郁症、癌症等。天然抗氧化剂能清除自由基且具有安全、健康等优点,受到人们广泛关注[23],如图9所示,在测定质量浓度为2~20 mg/mL时,清除率随样品质量浓度的增大而增大,当质量浓度达到20 mg/mL时,清除率达到了(17.97±0.47)%,表明DPoP对DPPH自由基有一定的清除能力。经拟合,DPoP曲线方程为y=0.02x2+1.33x-1.98,R2=0.994,IC50=31.03 mg/mL。
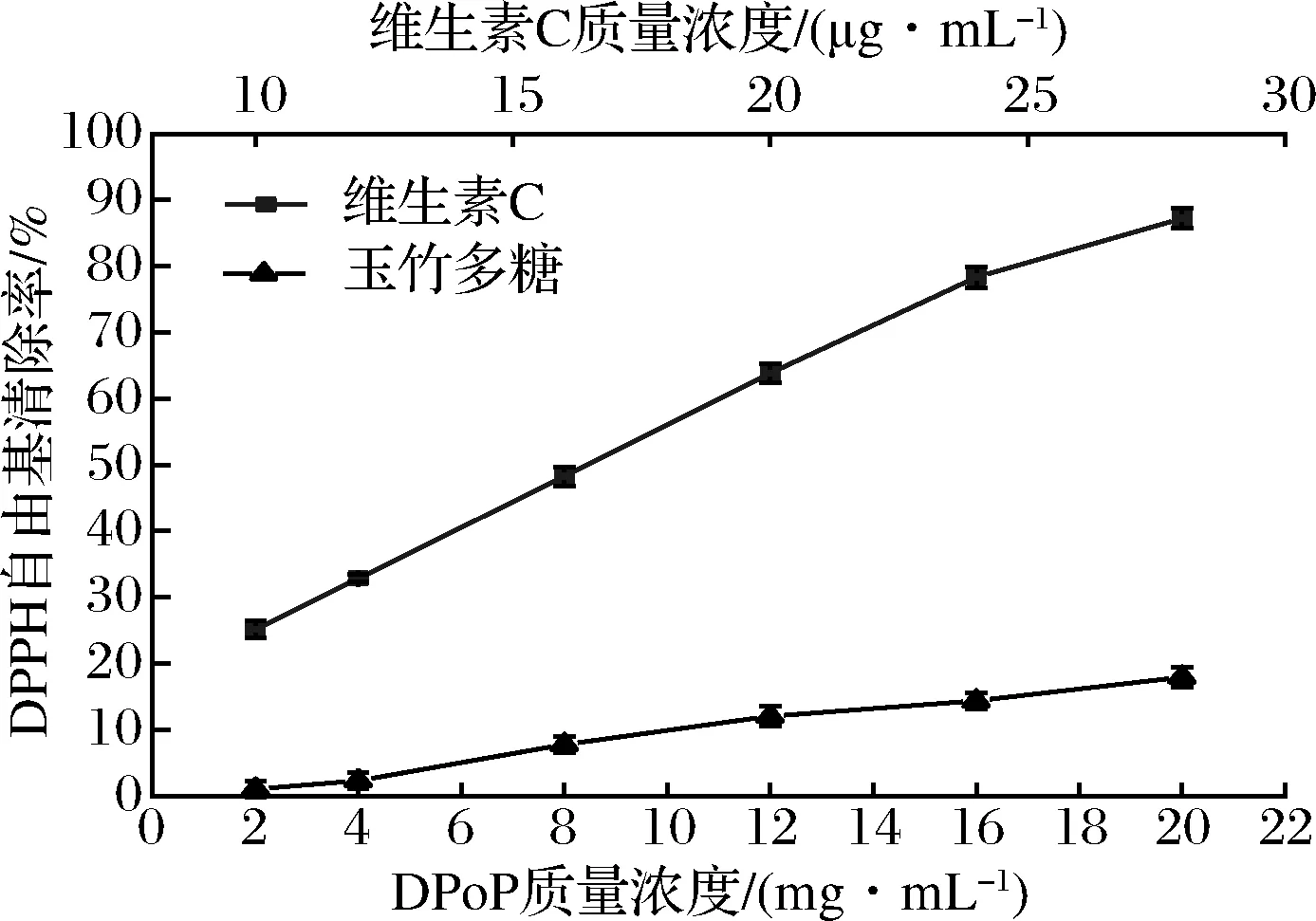
图9 DPoP对DPPH自由基清除率Fig.9 DPPH radical scavenging rate of DPoP
2.6 DPoP对ABTS阳离子自由基清除能力
ABTS阳离子自由基为稳定的有机自由基,在氧化剂作用下氧化成绿色的ABTS阳离子,当抗氧化物存在时,ABTS的产生会被抑制。如图10所示,结果表明DPoP对ABTS阳离子自由基有一定的清除作用,且在测定浓度范围内DPoP对ABTS阳离子自由基的清除效果具有浓度依赖性,当质量浓度达到20 mg/mL时,清除率达到了(40.69±0.34)%,经拟合,DPoP曲线方程为y=0.01x3-0.34x2+5.22x+0.38,R2=0.998,IC50=23.25 mg/mL。综合图9和图10,DPoP虽然具有一定的清除DPPH自由基和ABTS阳离子自由基的能力,但其清除作用均显著低于阳性对照维生素C,这可能是由于多糖的抗氧化机制与维生素C不同所致。多糖在体内是通过调节抗氧化酶的活性,利用抗氧化酶的还原作用将氧化物还原为低毒或无害的物质,从而达到清除自由基作用[24],玉竹多糖抗氧化机理有待进一步深入研究。
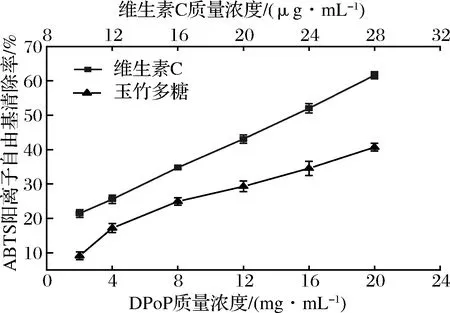
图10 DPoP对ABTS阳离子自由基清除率Fig.10 ABTS cationic radical scavenging rate of DPoP
2.7 DPoP的ORAC值
ORAC值最接近真实的生理环境氧化过程,具有可操作性强、灵敏度高等优势而被广泛应用于食品功能性成分抗氧化测定方面[10]。以水溶性维生素E为对照组,经计算得玉竹多糖的ORAC值为(121.67±3.65) μmol/g,DPoP在ORAC法中表现出较强的抗氧化能力。唐兰芳[7]测定低共熔溶剂提取DPsP的ORAC值也达到(130.53±8.29) μmol/g,与DPoP的ORAC值相近;唐仕荣等[25]通过超声辅助提取枸杞多糖,测得枸杞多糖的ORAC值为78.34 μmol/g,显著低于DPoP的ORAC值。图11为对照组标准曲线。
图11 荧光衰退标准曲线Fig.11 Fluorescent decay standard curve
2.8 DPoP抗糖基化能力
AGEs是糖基化反应的终末端产生的对人体有害物质,与糖尿病、心脏病、肾衰竭和老年痴呆等许多疾病的发展有关[16]。如图12所示,DPoP对BSA-Glu体系中糖基化终末产物AGEs的抑制作用仅稍低于氨基胍,表现出很强的抗糖基化能力。在测定范围内,DPoP对AGEs相对抑制率呈线性增长,当DPoP质量浓度达到2.0 mg/mL时,抑制率达到(87.63±0.66)%,展示出良好的抑制效果,作为从食物资源中提取的天然成分,DPoP必将具备很好的应用前景。

图12 DPoP对AGES相对抑制率Fig.12 Relative inhibition rate of AGES of DPoP
3 结论
DESs提取玉竹中的活性多糖较热水有更强的溶出能力,工艺确定以尿素-氯化胆碱体系作溶剂,采取响应面优化设计,得到最优提取条件:在尿素-氯化胆碱摩尔比为3∶1,提取温度92 ℃、提取时间41 min、DESs含水量19%,液料比29∶1的条件下,玉竹多糖最终提取率可达(29.03±0.54)%,为热水浸提法的4.67倍。
通过红外光谱分析确定DPoP性质与糖苷键类型,DPoP为酸性多糖,存在β-吡喃糖苷键与α-吡喃糖苷键。DPoP抗氧化测定结果均表明,DPoP具有抗氧化能力,DPoP对DPPH自由基、ABTS阳离子自由基清除能力随多糖质量浓度增大而增大,在质量浓度为20 mg/mL时,清除率分别达到(17.97±0.47)%、(40.69±0.34)%,ORAC值达到(121.67±3.65)μmol/g;糖基化终末端产物AGEs在体内积累是引发糖尿病的直接因素,抗糖基化结果表明,DPoP对AGEs的生成具有良好的抑制效果,当DPoP质量浓度达到2.0 mg/mL时,抑制率可达到(87.63±0.66)%,DPoP在新型降糖保健食品的开发方面有较大的应用潜力,有望进一步研究体内抗糖作用与机制以及进行相关功能性食品开发。
猜你喜欢
现代农村科技(2022年2期)2022-03-04
普洱学院学报(2021年6期)2022-01-14
复旦学报(医学版)(2021年5期)2021-10-13
文萃报·周二版(2020年24期)2020-06-26
艺术家(2020年12期)2020-03-02
癌症进展(2018年11期)2018-12-30
科技视界(2018年22期)2018-10-08
创作与评论(2017年7期)2017-05-20
文艺论坛(2017年7期)2017-05-16
中小企业管理与科技·下旬刊(2016年7期)2016-10-21
