
孟鲁司特钠咀嚼片在健康人体内的生物等效性
2022-04-29 00:55张小蕾王晨静李欣柳艳平高华曹玉
青岛大学学报(医学版) 2022年2期
张小蕾,王晨静,李欣,柳艳平,高华,曹玉
(青岛大学,山东 青岛 266021 1 药学院; 2 附属医院Ⅰ期临床研究中心)
孟鲁司特是一种新型的、强选择性的白三烯受体拮抗剂,其作用和其他白三烯受体拮抗剂类似(抑制炎症反应和渗出、抗过敏等),在过敏性鼻炎、多种哮喘、上呼吸道感染后咳嗽以及慢性阻塞性肺疾病等呼吸系统相关的疾病中应用广泛[1-7]。该药由默沙东制药有限公司研发,商品名为顺尔宁,1998年在美国上市,之后陆续在中国等许多国家销售使用。孟鲁司特的剂型包括片剂、咀嚼片和颗粒剂。其中,孟鲁司特钠咀嚼片服用方便,一天服用一次,安全性好,不良反应轻微,因此应用范围广,可用于成年人和12个月及以上儿童哮喘的预防和长期治疗,同时还能缓解季节性以及常年性过敏性鼻炎引起的不适。鉴于孟鲁司特的以上作用特点以及近些年来哮喘和过敏性鼻炎病人不断增多的趋势[8-9],通过生物等效性试验对孟鲁司特钠咀嚼片仿制药和原研药的一致性进行评价,可以确保仿制药在质量和疗效上与原研药一致,为后期在临床上以孟鲁司特钠咀嚼片仿制药替代原研药奠定坚实的基础,从而有效降低目标病人高额的医药费用[10]。对于通过一致性评价的药品国家也颁布了一系列的激励政策,如医保支付方面的支持、药品集中采购的倾斜、医疗机构采购与选用的优先权、对相应企业申请资金时给予的支持等[11]。在上述背景下,某药企研制了孟鲁司特钠咀嚼片仿制药,本研究在成年受试者空腹状态下观察上述仿制药和原研药的药代动力学参数及生物利用度,进而评价两种制剂的生物等效性,以期为仿制药上市提供技术支持。
1 材料与方法
1.1 仪器及软件
色谱系统包括:DGU-20A5R型脱气机、CBM-20A型控制器、LC-20AD型液相泵(日本岛津公司),CTC PAL型自动进样器(瑞士思特斯分析仪器有限公司)和DT-230A型柱温箱(上海楚越电子科技有限公司);串联质谱系统包括:API 4000型质谱仪、配套电喷雾离子源(美国Sciex公司);数据采集所用Analyst 1.6.2软件(美国Applied Biosystems公司)。
1.2 药品与试剂
受试制剂:孟鲁司特钠咀嚼片(国内某制药有限公司,规格为每片5 mg,批号为20170101);参比制剂:孟鲁司特钠咀嚼片,商品名为顺尔宁(默沙东制药有限公司,规格为每片5 mg,批号为M038748);孟鲁司特钠对照品(TLC Pharmaceutical Standards Ltd.,批号为1827-014A1,纯度为99.8%);内标:孟鲁司特钠-d6(Toronto Research Chemicals Inc.,批号为2-XAL-140-3,化学纯度为95%,核素纯度为99.5%)。以乙腈、甲醇、甲酸为色谱纯;水是自制的超纯水。
1.3 受试者选择
根据既往研究的药代动力学数据,孟鲁司特钠咀嚼片(5 mg)的个体内变异为17%。假定受试制剂和参比制剂的几何均值比为0.95~1.05,把握度为0.90,双向单侧显著性水平为0.05,个体内变异为17%,经统计软件计算得到需有20例受试者才可证明生物等效性。考虑到受试者脱落因素,本试验计划入组24例受试者。已入组的24例健康受试者,筛选期体格检查、实验室检查、生命体征评估、12导联心电图均正常或异常无临床意义。无受试者报告酗酒史、药物滥用史、献血/输血史。其中男性受试者20例,女性4例;平均年龄(26.33±5.31)岁;平均身高为(170.27±7.18)cm;平均体质量为(63.48±8.52)kg,平均体质量指数为(21.88±2.37)kg/m2。本研究已通过青岛大学附属医院医学伦理委员会的批准(药临伦审 QYFYEC 2017-019-01),所有受试者试验前均签署知情同意书。
1.4 试验方案
本试验采用随机、平衡、开放、单剂量、两周期、两序列、两交叉设计,清洗期为7 d。24例受试者按照签署知情同意书的先后顺序给予“筛选号”,参加体格筛选,筛选合格的受试者在第1次入住研究中心时获得“随机号”,并被随机分配到一个给药组。使用SAS 9.4统计软件编写随机化程序,按区组随机方法将受试者随机分为2组,每组12例。所有受试者于给药前1 d入住研究中心,每个周期内,受试者在空腹至少10 h后随机服用受试制剂或者参比制剂,用240 mL温水送服,给药前、后1 h内禁止饮水,分别于给药4、10 h后进食统一的清淡午餐和晚餐。试验过程中密切监测并记录受试者的生命体征和不良事件。经过7 d清洗期后,第2周期受试者按照方案规定交叉服用另一种药物,重复第1周期的流程。
每一周期每一受试者共采集16管静脉血(每管4 mL),分别在给药0时(给药前1 h内)以及给药0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0、6.0、8.0、10.0、12.0、24.0 h后采集,采集的血样置于事先贴有标签的真空采血管中。血样采集后在预冷离心机中(2~8 ℃)以3 000 r/min离心10 min,分离血浆,取800 μL血浆放入测试管中,剩余血浆放入备份管中。密集点血浆样本暂存于-20 ℃冰箱中,于24 h内转移至(-70±10)℃冰箱中冻存,非密集点血浆样本直接放(-70±10)℃冰箱中冻存,以供药代动力学分析[12]。
1.5 色谱与质谱条件
色谱柱为:Phenomenex Luna C18(50 mm×2 mm,5 μm),无预柱;流动相:水-乙腈,采用梯度洗脱(0 min:水-乙腈(25∶75);2.0 min:水-乙腈(25∶75);2.1 min:水-乙腈(5∶95);2.9 min:水-乙腈(5∶95);3.0 min:水-乙腈(25∶75);4.0 min:水-乙腈(25∶75));流量:0.45 mL/min;柱温:40 ℃;进样量:20 μL。
采用电喷雾离子化技术、正离子模式、多反应监测的液相色谱-串联质谱法进行分析。离子源参数如下:气帘气(CUR)压力206.85 kPa,喷雾电压5 000 V,温度550 ℃,离子源气体1(GS1)压力为344.75 kPa,离子源气体2(GS2)压力344.75 kPa。孟鲁司特监测离子对为m/z586.2→m/z422.2,去簇电压(DP)为77 V;内标孟鲁司特-d6离子对为m/z592.3→m/z427.2,DP为77V;扫描时间为200 ms。
1.6 血浆样品处理
取样本50 μL加到96孔聚丙烯板中,加入100 μg/L内标工作溶液50 μL,在室温黄光条件下振摇样本板约1 min后,加入乙腈300 μL,于室温黄光条件下振摇混匀约10 min。再将上述样本板于4 ℃条件下,以4 000 r/min离心10 min,转移上清液200 μL至另一干净的96孔聚丙烯板中,并加水50 μL进行稀释,在室温黄光条件下振摇混匀约10 min。最后将该样本板于4 ℃条件下,以4 000 r/min离心5 min,取上清液,进样20 μL进行分析。
1.7 方法学验证
1.7.1方法选择性 取6份不同来源的空白人血浆和相应空白血浆制备的浓度为2 μg/L的定量下限样本,用以评价该分析方法的选择性。待测物通道的干扰峰面积不超过相应基质制备的定量下限样本中待测物峰面积的20%,内标通道的干扰峰面积不超过相应基质制备的定量下限样本中内标峰面积的5%。
1.7.2标准曲线与定量下限 应用空白人血浆新鲜制备8个浓度水平(2、4、10、50、200、600、900、1 000 μg/L)的标准曲线样本,按“1.6血浆样品处理”中方法处理后进样分析。
1.7.3精密度与回收率 用空白人血浆新鲜制备5个浓度水平(2、6、40、500、800 μg/L)的质控样本,按“1.6血浆样品处理”中方法处理后进样测定。每个浓度水平的质控样本各6份在同一天内进行测定,连续测定3 d,以此计算该测定方法的批内与批间精密度。
取空白人血浆,提取后向提取物中添加待测物和内标制得浓度分别为6、500和800 μg/L的参比样本。再应用空白人血浆配制浓度分别为6、500、800 μg/L的质控样本作为测试样本。每个浓度水平的参比样本和测试样本分别各配制6份。计算内标校正后的提取回收率(每个浓度水平的测试样本中待测物与其内标峰面积比的均值除以同浓度参比样本中待测物与其内标峰面积比的均值),用以评估该分析方法的提取回收率。
1.7.4稳定性试验 对于待测物在全血和血浆样本中的稳定性,均观察低、高(6、800 μg/L)两个浓度水平的质控样本,每个浓度水平的样本各6份,分别进行全血样本室温白光放置2.0 h、血浆样本室温白光放置2.5 h、血浆样本室温黄光放置19.5 h、血浆样本-80 ℃循环冻融5次、血浆样本-80 ℃放置36 d的稳定性试验。
1.8 数据处理及统计分析
使用WinNonlin®7.0软件,按照非房室模型的参数计算方法,计算孟鲁司特钠的人体药代动力学参数,并采用SAS 9.4统计软件对所得数据进行统计学分析。受试制剂与参比制剂的达峰时间(Tmax)比较采用配对样本的非参数检验。最大血药浓度(Cmax)、从0时到最后计算时间点浓度-时间曲线下面积(AUC0-t)、从0时到无限时间浓度-时间曲线下面积(AUC0-∞)的数据在进行自然对数转换以后,进行方差分析(ANOVA)及双向单侧t检验,计算受试制剂和参比制剂血浆中孟鲁司特Cmax、AUC0-t和AUC0-∞几何均值比的90%置信区间,进行生物等效性评价。若受试制剂与参比制剂药代动力学参数(Cmax、AUC0-t、AUC0-∞)几何均值比的90%置信区间在80.00%~125.00%范围内(包括边界值),则可认为受试制剂与参比制剂等效[12]。
2 结 果
2.1 方法学验证
方法学验证结果显示,空白人血浆中的内源性物质对孟鲁司特钠和内标的测定没有显著性影响,该方法的选择性良好。以孟鲁司特钠的质量浓度为横坐标(x),待测物与内标峰面积比值为纵坐标(y)进行线性回归,所得回归方程如下:y=0.007 15x+0.000 38(r=0.999 00),权重因子为1/x2,线性范围为2~1 000 μg/L,最低定量限为2 μg/L。在不同试验条件下,孟鲁司特钠全血和血浆样本均保持稳定且相对标准偏差(RSD)<15%,符合方法学的要求。精密度与回收率试验结果见表1。
2.2 药代动力学参数
入组的24例受试者分别空腹口服孟鲁司特钠受试制剂和参比制剂5 mg后的主要药代动力学参数见表2。由表2数据可知,两种制剂中,孟鲁司特钠的主要药代动力学参数基本相似,且与国外有关文献结果一致[13-14]。

表1 精密度与回收率试验结果
2.3 生物等效性分析
受试制剂和参比制剂的主要药代动力学参数Cmax、AUC0-t及AUC0-∞在进行对数转换后,进行交叉试验设计的方差分析。分析结果显示,孟鲁司特钠咀嚼片Cmax制剂间的差异有统计学意义(F=6.211,P<0.05),但不影响受试制剂的生物等效性评价,给药序列间、给药周期间的差异均无统计学意义(P>0.05);AUC0-t及AUC0-∞在给药序列间、制剂间和给药周期间的差异也均无统计学意义(P>0.05)。两种制剂的Tmax比较差异也无统计学意义。采用双向单侧t检验,计算得出受试制剂与参比制剂Cmax、AUC0-t和AUC0-∞几何均值比的90%置信区间分别为81.03%~96.20%、89.78%~102.69%和89.99%~103.12%,均符合药物制剂人体生物利用度和生物等效性试验指导原则中生物等效性的等效范围要求(80.00%~125.00%)。以上数据表明,在空腹状态下受试制剂和参比制剂生物等效。
2.4 安全性评价
本研究的24例受试者在试验期间共有3例受试者发生4次不良事件,分别为第5号受试者第2周期服参比制剂后发生上呼吸道感染;第9号受试者第2周期服参比制剂后发生T波改变、窦性心动过速;第17号受试者在第2周期服参比制剂前发生晕厥。其中,前2例受试者发生的3次不良事件均为轻度,无需处理;后1例受试者发生晕厥为中度不良事件,给予口服500 g/L葡萄糖液后好转。经分析,前2例受试者发生的3次不良事件与研究药物可能有关;而后1例受试者发生晕厥与研究药物无关,因为17号受试者出现晕厥时,距离第1周期服药已经7 d,尚未服用第2周期的药物,且已空腹10 h,口服葡萄糖后完全好转无后遗症,持续时间共14 min,因此判定晕厥为低糖血症所致。整个试验过程中未观察到严重或导致退出的不良事件。同时,生命体征和体格检查也均未出现有临床意义的异常结果。故孟鲁司特钠咀嚼片受试制剂与参比制剂的安全性和耐受性良好。
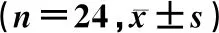
表2 两种制剂主要药代动力学参数比较
3 讨 论
孟鲁司特钠口服即可有效,吸收迅速,无论单独服用还是与其他药物联合使用,临床疗效显著;此外,因毒性小,不良反应轻微,病人的耐受性和依从性都比较高,而且孟鲁司特钠适用于儿童和成年目标病人,故该药自2002年在国内上市以来,临床应用日趋广泛[15]。
本研究采用高效液相色谱-质谱联用法测定血浆中孟鲁司特钠的血药浓度,经方法学验证,该方法选择性、灵敏度、稳定性、精密度和回收率均符合要求,待测药物浓度与响应值的线性关系良好。
本次试验方案的设计主要参考国家药品监督管理总局2016年颁布的《以药动学参数为终点评价指标的化学药物仿制药人体生物等效性研究技术指导原则》[12]。按照该指导原则,将原研药顺尔宁作为本研究的参比制剂。同时,该指导原则中明确规定,试验给药之间应有足够长的清洗期(一般为待测物7倍半衰期以上),孟鲁司特的半衰期为2.7~5.5 h,因此本研究在两次给药之间设置了7 d的清洗期。本研究采用单剂量、双周期、两序列的交叉试验设计,每1例入组的受试者均连续服用两次不同的药物制剂,等同于自身对照,从而减少了不同试验周期以及个体差异对结果的干扰。另外,为了减少与药品间差异无关的因素的影响,试验纳入健康的男性和女性受试者,最终,所有受试者均完成了整个研究,并且均按照要求用药,各周期服药依从性良好。
本文研究结果显示,受试制剂与参比制剂中,孟鲁司特钠的主要药代动力学参数基本相似,且与已有的文献报道结果一致[13-14]。两种制剂的平均血药浓度-时间曲线的走向吻合度也比较高,说明二者在人体内的药代动力学过程接近。进一步的生物等效性分析结果表明,两种孟鲁司特钠咀嚼片在空腹状态下吸收速度和程度相当,符合生物等效性的判定标准,而且两种制剂的安全性和耐受性较好。
综上所述,在空腹状态下,孟鲁司特钠咀嚼片受试制剂与参比制剂具有生物等效性。
猜你喜欢
化工管理(2022年14期)2022-12-02
药学教育(2022年3期)2022-07-09
中国典型病例大全(2022年12期)2022-05-13
中国典型病例大全(2022年12期)2022-05-13
中国典型病例大全(2022年13期)2022-05-10
中国典型病例大全(2022年9期)2022-04-19
药学研究(2021年5期)2021-11-29
医学概论(2021年18期)2021-01-21
分析化学(2014年12期)2014-12-18
