
磷化钴(111)面析氢性能的第一性原理
2022-04-27 08:28段升权高洪涛
青岛科技大学学报(自然科学版) 2022年2期
段升权,高洪涛
(青岛科技大学 化学与分子工程学院,山东 青岛 266042)
电解水制氢是解决环境污染、能源短缺的有效途径。迄今为止,铂(Pt)仍是析氢反应(HER)中最有效的电催化剂[1]。然而Pt成本高,易失活,阻碍了其在工业规模上的广泛应用。过渡金属磷化物(TMP)因其具有相对其他非贵金属电催化剂较高的催化活性,且与铂相比较低成本而成为电催化剂的替代材料[2]。
磷化钴(CoP)是一种典型的TMP,不同形态的CoP通常会暴露出不同的晶面,从而导致表面上的原子排列不同,实验中可以通过不同几何结构(如纳米颗粒[3]、纳米棒[4]、微晶片[5]以及多孔结构[6]等)的特定平面来提高CoP的催化性能,但它们的活性部位和稳定性尚不清楚[7]。
在众多的纳米CoP结构中,CoP(111)面具有较好催化活性,研究中关注也最多[8],但是对于H2吸附脱附电催化机理仍缺乏深入理解。本研究基于DFT的第一性原理对CoP(111)表面活性进行探究,探讨了最稳定(111)表面原子分布对表面析氢活性的影响,并尝试采用掺杂Os、Mn等不同原子提高表面活性,通过电荷分布和态密度分析掺杂对CoP析氢催化活性的影响。
1 计算方法
密度泛函理论(DFT)计算通过VASP软件来实现[9],使用广义梯度近似GGA中的PBE方法作为电子交联相关函数,采用PAW函数描述原子和原子核之间的相互作用,平面波的截断能设置为450 e V。当原子间每个离子的受力小于0.2 e V·nm-1,总的能量变化小于10-5e V时弛豫结束。K点网格选取基于Gamma方法采取6×5×1分布。为了尽可能减小表面模型之间的相互作用,沿着Z轴的真空层取1.5 nm。
可以采用表面能[10](如下式所示)来确定CoP(111)面最稳定的表面结构(s),表面能越小,结构越稳定。

HER活性和电催化剂表面对氢的吸附能密切联系,在催化剂表面氢吸附能ΔEH可以根据公式(2)计算
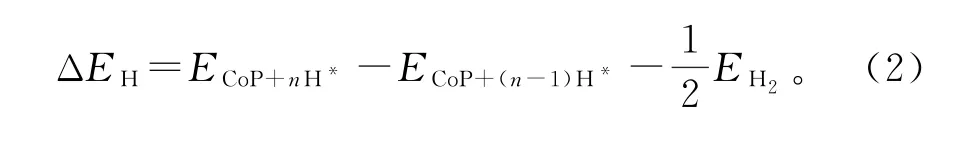
其中,ECoP+nH*和ECoP+(n-1)H*代表吸附n和n-1个氢质子的CoP的总能量。EH2是气相H2分子的总能量。吸附H的吉布斯自由能由以下公式得到
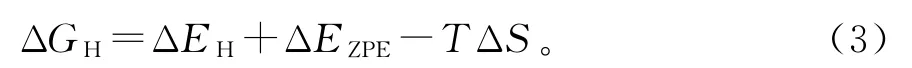
其中,ΔEZPE和ΔS分别代表标准条件下吸附H*和气相H2之间的零点能量变化和熵变化,T表示温度(298.15 K)。ΔS近似等于,而SH2是标准条件下气相中分离出的H2分子的熵。ΔEZPE可以表示为
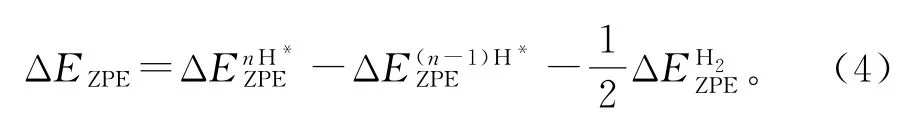
2 结果与讨论
2.1 CoP的体相结构和(111)面表面结构
体相CoP(Pnma)结构中,Co3+与6个等效的P3-原子键合,形成扭曲的CoP6八面体构型(图1),P3-被6个当量的Co3+原子包围,形成高度扭曲的三角棱镜。体相CoP(a=0.506 7 nm,b=0.326 3 nm,c=0.554 5 nm)的优化后晶胞参数与实验数据高度一致(a=0.507 7 nm,b=0.328 1 nm,c=0.558 7 nm)[12]。说明作者采用的计算方法可行,计算结果是可信的。
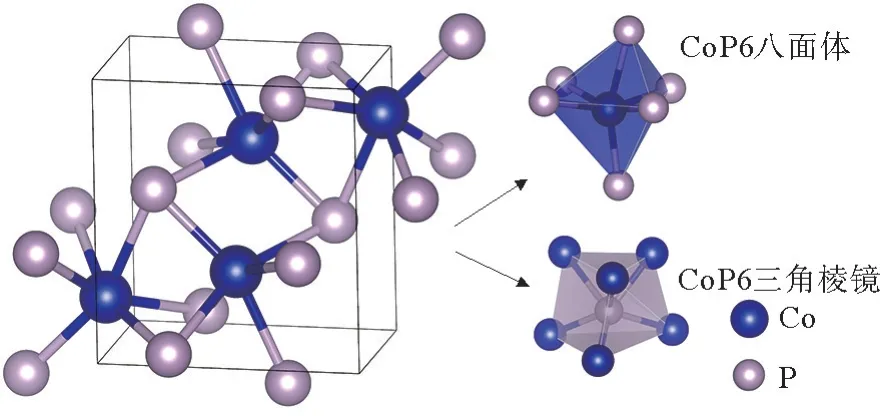
图1 CoP体相结构Fig.1 Bulk structure of CoP
本研究选取CoP(111)面来研究氢原子在其表面的吸附。为了模拟各种复杂分子面的HER电催化活性,需要稳定的(111)面的模型。CoP(111)面有8个不同的原子分布结构,使得表面暴露出不同的原子构型,在图2中用横线表示,这些不同的原子分布用数字1~8表示,表面9~16基本为表面1~8的对称面。因此,只需研究表面1~8分布结构的稳定性即可。
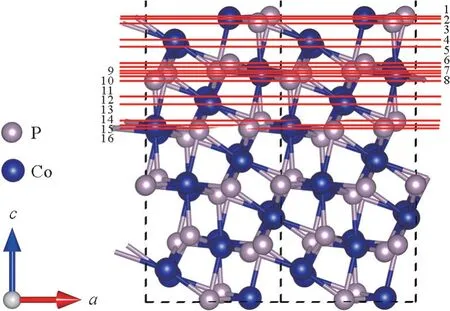
图2 CoP(111)晶面表面分布原子状况Fig.2 CoP(111)crystal plane surface exposed atomic conditions
表面1~8不同原子构型和原子相对位置的表面能见表1。表面1时具有最大表面能2.329 J·m-2,表面构型为表面5时,具有最低表面能为1.211 J·m-2,其平衡构型见图3。表面5构型与表面1构型相比有两个Co原子会暴露在外表面,另外两个Co原子与四个P原子处于比较接近的水平面,通过驰豫,发现表面Co原子位置均沿Z方向向下移动,Co1原子经历最大的向下弛豫,为0.044 nm;Co2原子次之,向下位移0.036 nm,而表面P原子在Z方向位移较小,这表明CoP(111)晶面上的Co原子可以向内部收敛,降低表面能。从图2可以看出表面1~3和6~8彼此间距较小,原子排列密集,表面4~5与其他表面间距较大,原子排列较为疏松,当(111)晶面以密集表面为表面构型时表面能较大,原因是需要更多的能量来破坏强共价的Co-P键。而当(111)晶面以疏松表面为表面构型,这种结构的表面能偏低,由此可以得出结论,(111)晶面暴露出含Co原子的疏松表面是最稳定的,也是最有可能存在于环境中的情况。因此,选择(111)晶面最稳定的表面5上进行了以下吸附H计算[13]。

表1 表面1~8不同原子构型的表面能Table 1 Surface 1—8 exposed surface energy of different atomic configurations
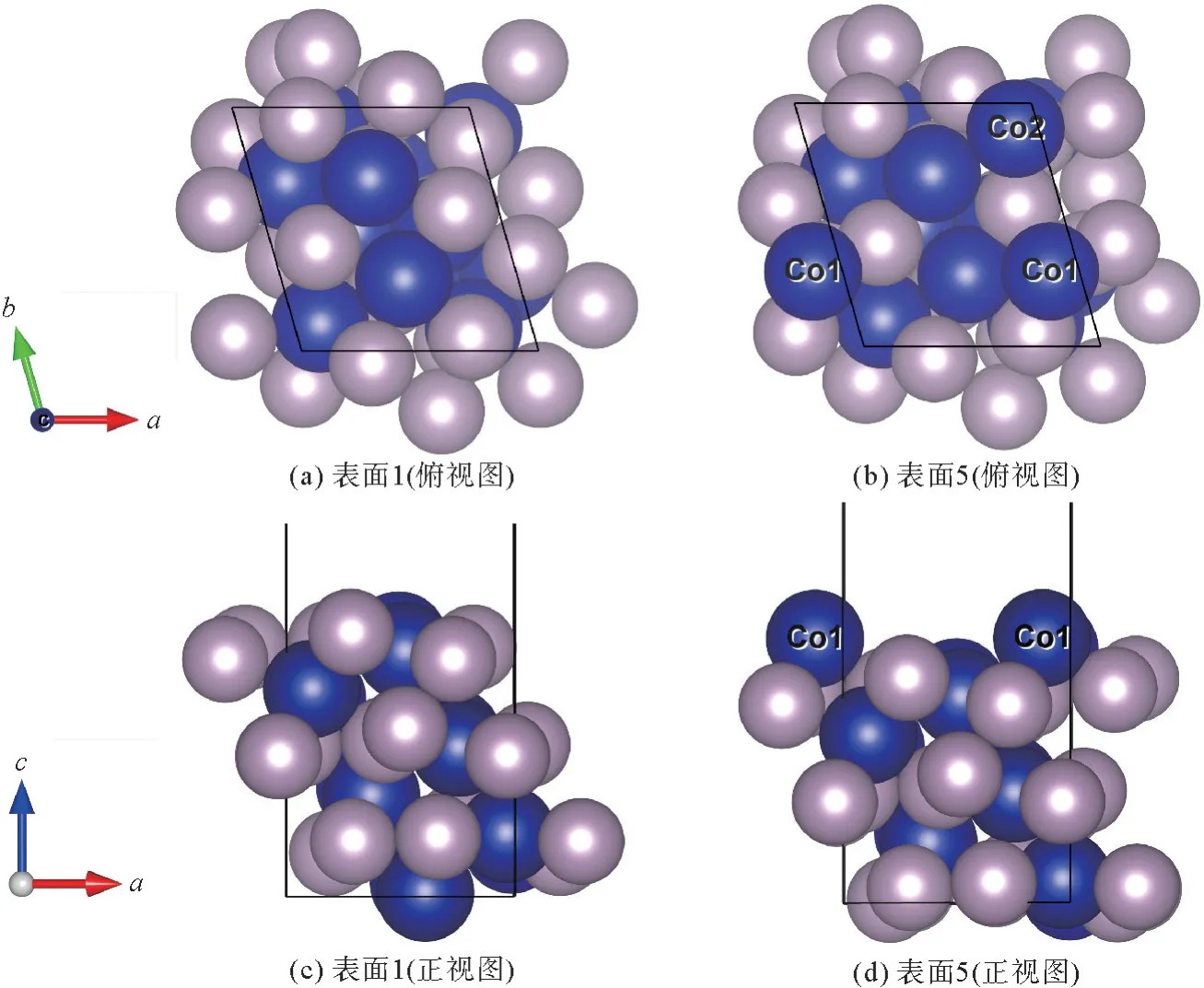
图3 CoP(111)表面1和表面5的俯视图和正视图Fig.3 Top view and front view of CoP(111)surface 1 and surface 5
2.2 CoP(111)面的析氢性能研究
HER活性通常用吉布斯吸附自由能ΔGH来描述,ΔGH越接近于零说明电催化剂HER活性越好[14]。图4显示了具有不同氢覆盖率的CoP(111)的优化结构。CoP(111)上有两种稳定的氢吸附位点(即表面的钴顶部和钴桥位点),最多可以吸附8个H原子,使H的覆盖范围在1/8~8/8范围内变化,这些吸附位置也符合CoP的结构,P原子与其他原子之间的距离太长,无法在H原子吸附过程中形成桥键[15]。图5显示了CoP(111)在不同氢覆盖率(此处为100%覆盖率=21个·nm-2)下计算出的ΔGH和ΔEH。对于前1/8覆盖度的氢原子,它们被强吸附在钴桥位点。在2/8~5/8的氢覆盖率期间,主要吸附在在钴顶部位点,导致钴原子上有一个以上的氢原子,更重要的是,在2/8~3/8和5/8这个范围内ΔGH接近于零,这表明在该覆盖度范围内CoP具有较好的HER催化活性,因此钴顶部位点是(111)表面HER的最佳活性位置。对于6/8至8/8的覆盖率,氢原子被吸附在其他钴桥位点(在CoP(111)表面3/8(H3)和5/8(H5)覆盖度下会出现轻微的能量波动,这是由吸附的氢原子之间的相互作用引起的)[16]。
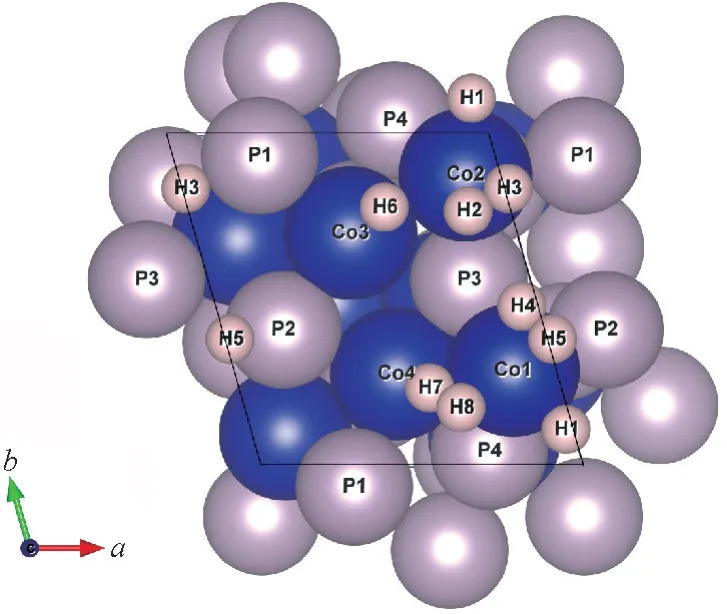
图4 CoP(111)表面吸附H原子位点示意图Fig.4 CoP(111)surface adsorption H atom site
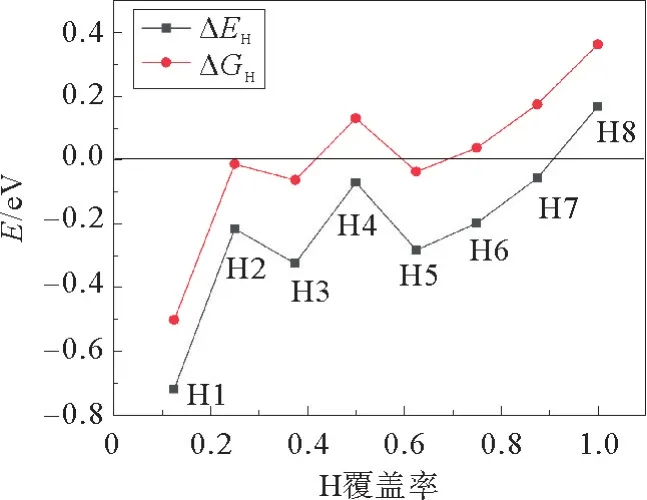
图5 CoP(111)表面吸附H原子位点对应的吸附吉布斯自由能Fig.5 Gibbs free energy of adsorption corresponding to H atom site adsorbed on CoP(111)surface
根据d带中心理论,接近费米能级的d带中心显示出较强的吸附,而远离费米能级的d带中心显示出较弱的吸附。通过表面Co原子的分态密度(PDOS,图6)可以来解释不同Co原子吸附能力的差异。因为Co2原子的d带中心(-126.281 kJ·mol-1)高于Co1原子的d带中心(-127.339 kJ·mol-1),因此Co2原子对H原子的吸附要强于Co1原子。Co3原子的d带中心(-180.266 kJ·mol-1)和Co4原子的d带中心(-169.149 kJ·mol-1)远小于Co1和Co2,使得H原子优先吸附在Co1和Co2位点,Co3、Co4和Co1、Co2原子之间较大的d带差异可能是由于它们周围的不同几何结构引起的。因此,将Co1、Co2和Co3、Co4原子分别标记为有效和无效位点。
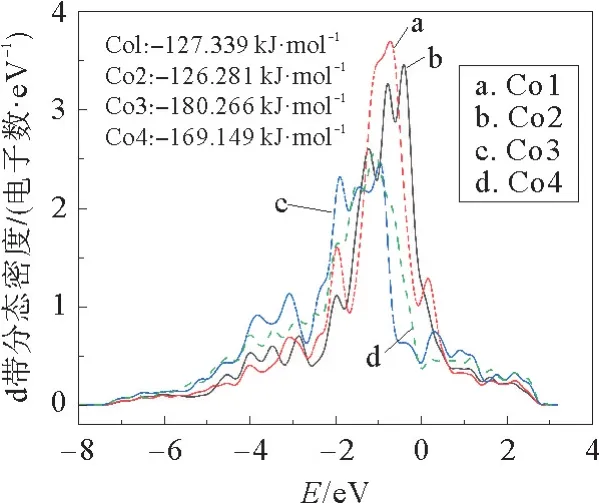
图6 CoP(111)表面的部分状态密度(PDOS)Fig.6 Partial state density(PDOS)of CoP(111)surface
2.3 在CoP(111)表面上的掺杂改善
掺杂是改善材料表面物理和化学性质的有效方法,可以改变表面的电子环境[17]。为了进一步提高CoP(111)表面的析氢反应活性,研究了在无效位点Co4取代掺杂金属元素原子来改变邻近Co1位点电子环境,以此降低H4的吉布斯自由能,增加有效析氢位点,提高HER活性。由于Co元素处在元素周期表3d金属周期内,周期内的大多数元素半径接近,掺杂同周期的3d金属元素有可能改变CoP的HER性质,而且这些元素通过掺杂改性也有可能在实验上真正实现。这里通过掺杂Cr、Fe、Ni、Cu等和Co半径接近的原子以及贵金属原子来改性表面HER活性。文献[17]表明掺杂浓度<5%可以避免晶格变化,本研究选择掺杂浓度为4.2%这个典型且合适的浓度。当然也对不同浓度掺杂和表面其他Co原子位点进行了掺杂取代,效果不尽如意。掺杂后,没有引起表面可见的结构性变化,计算掺杂体系的ΔGH见表2。根据H吸附吉布斯自由能的变化,可以看出替换掺杂Fe、Ni、Cu、Pd、Ag、Zn原子没有有效降低H4的ΔGH值,掺杂Mo、Cd、Ti、Rh、Ru原子可以有效降低H4的ΔGH值,但H2、H3和H5的H吸附吉布斯自由能值也开始大于0,说明掺杂Rh、Ru原子对其他H吸附位点也产生了影响;替换掺杂Mn、Os原子在Co4位点可以有效降低H4的吸附能,且对其他H吸附位点没有太大影响。更加详细的探索了Mn、Rh、Os和Ru原子的掺杂对HER催化活性的影响,H吸附吉布斯自由能见图7。
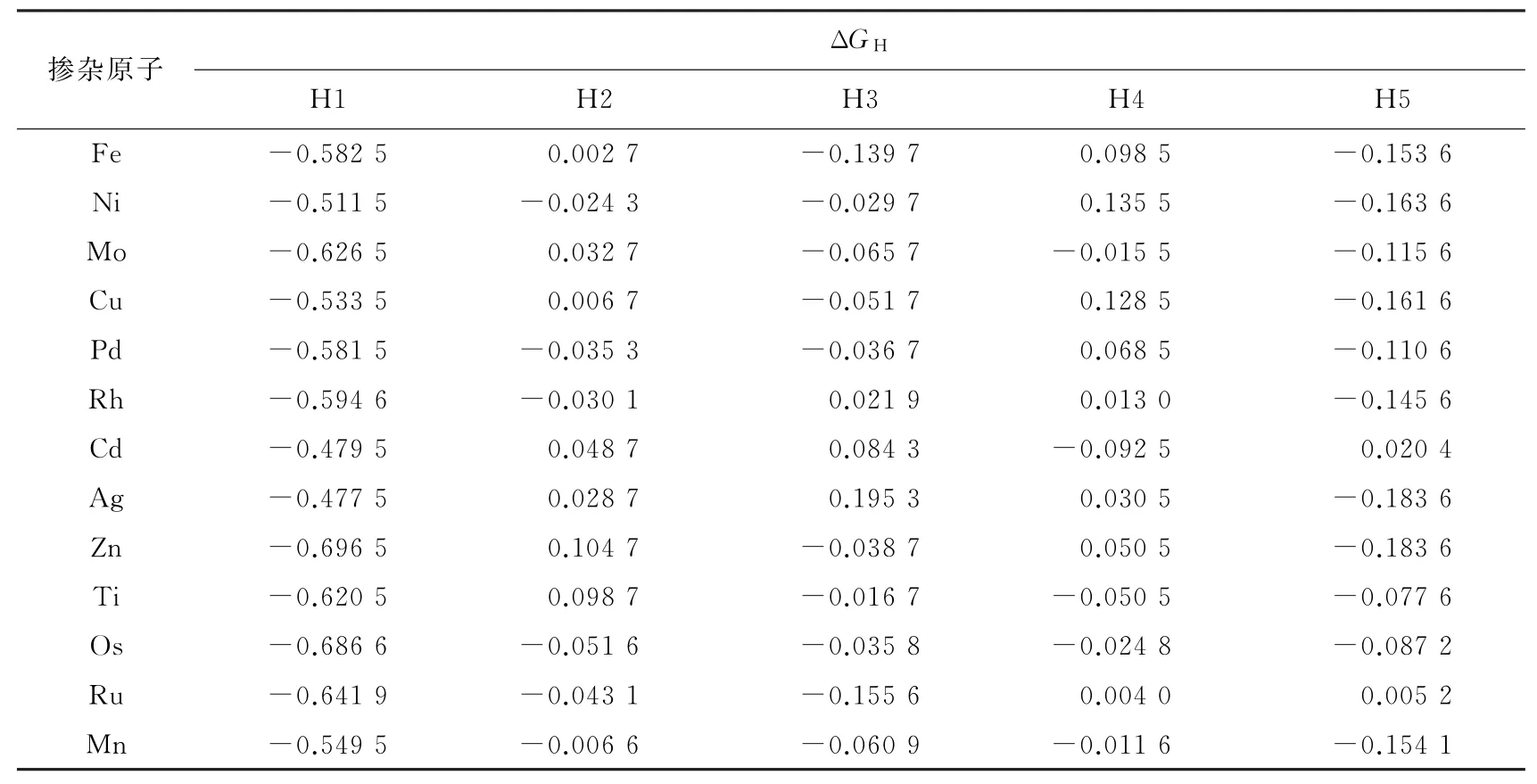
表2 替换掺杂Co4原子对CoP(111)表面HER的影响Table 2 Effect of replacing doped Co4 atoms on the HER of CoP(111)surface
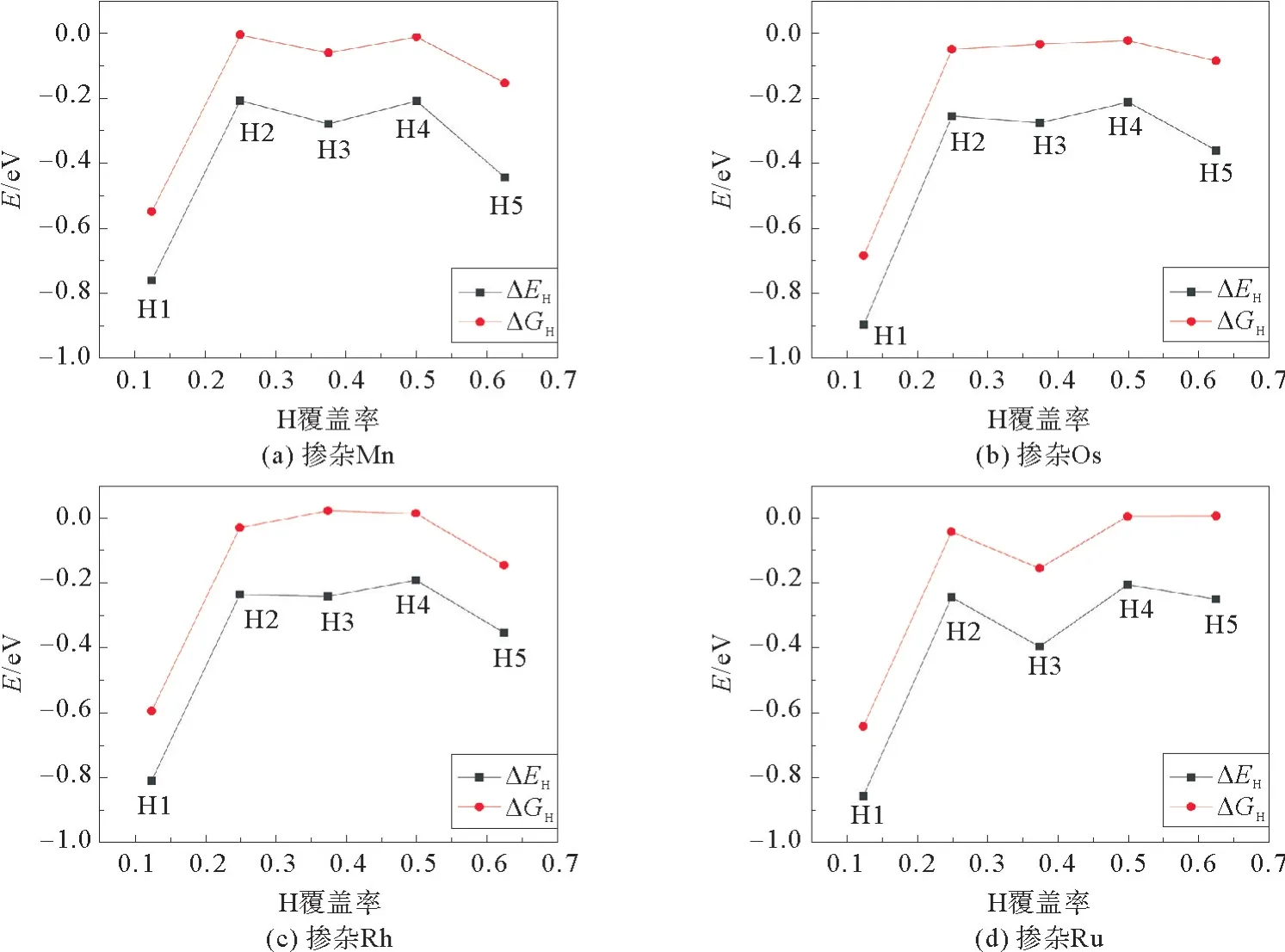
图7 掺杂Mn,Os,Rh,Ru元素的CoP(111)晶面的H吸附吉布斯自由能图Fig.7 Gibbs free energy diagram of H adsorption on CoP(111)crystal plane doped with Mn,Os,Rh and Ru elements
CoP(111)表面掺杂Mn、Rh、Os和Ru原子后,Co1原子的d能带中心增大,如图8所示。根据表3所示的H4吸附前Co1电荷分布数据,可以通过掺杂元素的电负性比Co的电负性弱来解释d带中心的增加,Bader电荷表明Co4位点取代掺杂元素使得Co1氧化数增加,损失更多的电子,电子云密度下降,这将有利于H原子的吸附。Mn、Os元素的掺杂增强了Co1的H吸附能力,整体提高了HER有效活性范围。此外,Os的5d轨道使得Co1 DOS向低能区移动,说明Os的掺杂促使Co1电子向导带转移,这能促进H原子的吸附和析氢反应的进行。
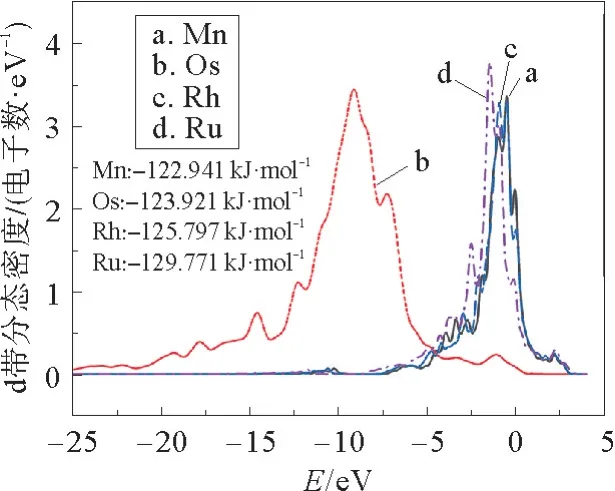
图8 对Co4原子取代掺杂后Co1原子的PDOS图及d带中心值Fig.8 PDOS diagram and d-band center value of Co1 atom after substitution and doping of Co4 atom

表3 Co4原子被取代时Co1的价电荷Table 3 Valence charge of Co1 when Co4 atom is substituted
3 结 论
采用第一性原理计算方法研究了CoP(111)表面不同原子分布对表面析氢活性的影响,并考察了原子掺杂提高表面析氢活性的可能性。结果表明(111)晶面Co原子在外表面的原子分布是最稳定的,具有最低表面能1.211 J·m-2,也是最有可能存在于环境中的情况;此外CoP(111)面在2/8~3/8 H覆盖度下ΔGH接近于零,这表明在该覆盖度下CoP具有较好的HER催化活性,表面的Co2顶点为主要活性位点;掺杂Mn,Os原子后改变了CoP(111)面的电子环境,H4原子吸附吉布斯自由能会下降接近负零,使得Co1位点得到有效利用,析氢活性位点的数量增加,析氢反应最优活性范围增加到2/8~5/8,提高了HER性能,表明表面原子分布对表面性能有很大影响,表面活性可以通过掺杂来改善。
猜你喜欢
分子催化(2022年1期)2022-11-02
分析测试学报(2022年9期)2022-09-21
中国农业科学(2022年16期)2022-09-19
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
装备维修技术(2020年5期)2020-11-20
科技传播(2019年22期)2020-01-14
电脑知识与技术(2018年19期)2018-11-01
