
Niche Differentiation of Phenol-Degrading Microorganisms in UASB Granular Sludge as Revealed by Fluorescence in situ Hybridization
2022-04-22 11:42KengoKuotKeiIgrshiMsyoshiYmdYsuyukiTkemurYuYouLiHidekiHrdc
Engineering 2022年2期
Kengo Kuot*, Kei Igrshi Msyoshi Ymd, Ysuyuki Tkemur Yu-You Li Hideki Hrdc
a Department of Civil and Environmental Engineering, Tohoku University, Sendai 980-8579, Japan
b Department of Urban Environmental Design and Engineering, National Institute of Technology, Kagoshima College, Kirishima 899-5193, Japan
c New Industry Creation Hatchery Center, Tohoku University, Sendai 980-8579, Japan
Keywords:Cryptanaerobacter Fluorescence in situ hybridization Anaerobic phenol degradation Syntrophorhabdaceae Syntrophus UASB granular sludge
ABSTRACT A microbial community structure of granules harvested from an anaerobic sludge blanket reactor treating phenolic wastewater was investigated using fluorescence in situ hybridization (FISH) and clone library construction. Clones of Syntrophorhabdaceae and Cryptanaerobacter were observed to be responsible for phenol degradation. For accurate taxonomic assignment of Cryptanaerobacter clones, phylogenetic analysis using nearly full-length 16S ribosomal RNA (rRNA) gene sequences was necessary. Three oligonucleotide probes were designed to detect the following three taxonomic groups:Syntrophorhabdaceae, Cryptanaerobacter, and Syntrophus. FISH analysis of thin sections of anaerobic granules showed a random distribution of bacteria and archaea. However, a well-defined distribution of Syntrophorhabdaceae, Cryptanaerobacter, and Syntrophus was observed. Cryptanaerobacter and Syntrophus were found on the outer layer of the granules and were closely associated with each other,while Syntrophorhabdaceae was located in the deeper part of the granules. Such specific distribution of the bacteria is most likely due to their metabolic association and affinity for the substrate. Phenol degradation in the granular sludge was observed to be carried out in the following way. First,Cryptanaerobacter converts phenol to benzoate, which is then degraded by Syntrophus into acetate.This syntrophic degradation of phenol occurs near the surface of the granule, where the phenol concentration is high. In the deeper part of the granule, where the phenol concentration is lower,Syntrophorhabdaceae degrades phenol into acetate.We observed that Syntrophorhabdaceae is less likely to produce benzoate as an intermediate to feed the neighboring organisms, which contradicts the theories presented by previous studies.
1. Introduction
Phenol is a widely used and industrially important chemical compound. As a result, phenol is also a common contaminant in the wastewater produced by industries such as resin manufacturing, coal gasification, oil refining, mining, and more [1]. The high solubility of phenol in water increases its mobility and causes widespread contamination of the environment. Several technologies have been developed, tested, and implemented for the degradation and removal of phenol from various types of wastewater[1-3]. Anaerobic biological degradation of phenol is an effective treatment technology because of its ability to completely degrade phenol, its low energy consumption, and its potential for generating bioenergy in the form of methane gas. Anaerobic microbial consortia degrading phenol have been extensively studied and investigated [2,4-9]. However, a systematic study of the microorganisms participating in phenol degradation and their roles is lacking.
It has been reported that the major groups of microorganisms participating in phenol degradation are Syntrophorhabdaceae,
Pelotomaculum, Desulfotomaculum, Syntrophus, and Clostridium
[2,7,9,10]. There are also mixed accounts on the roles of various other microorganisms in phenol biodegradation.Syntrophorhabdus aromaticivorans (S. aromaticivorans) [11] and Cryptanaerobacter phenolicus(C. phenolicus) [12] are two known species that can utilize phenol for growth in methanogenic environments.Many studies have described the phenol degradation pathway as phenol being degraded to acetate by Syntrophorhabdus in syntrophic association with hydrogenotrophic methanogens. These studies have also suggested that phenol is first converted to benzoate by Syntrophorhabdus or Pelotomaculum, and is then converted to acetate by Pelotomaculum, Desulfotomaculum, or Syntrophus [4,5,8,13-15].However,there has been no definitive evidence suggesting phenol utilization by any pure culture of Pelotomaculum in any published report.Therefore,it is less likely that Pelotomaculum converts phenol to benzoate. Also, it is less likely that Syntrophorhabdus could be a benzoate producer for other benzoate scavengers, because the conversion of phenol to benzoyl-coenzyme A (CoA) by S. aromaticivorans is an adenosine triphosphate (ATP)-consuming process. S. aromaticivorans needs to further degrade benzoyl-CoA to obtain energy[16].This reasoning gives rise to two key questions:① If Syntrophorhabdus produces benzoate as an intermediate,which then is consumed by other microorganisms, how does Syntrophorhabdus thermodynamically benefit from this process?② If the syntrophic microorganisms, such as Syntrophus and Pelotomaculum, do not feed on the intermediates produced by Syntrophorhabdus, what is the major substrate allowing them to maintain their abundance in the phenol-degrading consortium?
To answer these questions,we investigated the microbial community structure of a granular sludge sampled from an up-flow anaerobic sludge blanket(UASB)reactor treating phenol.After analyzing the microbial community structure by constructing clone libraries of 16S ribosomal RNA (rRNA) gene sequences, fluorescence in situ hybridization (FISH) with newly designed probes was applied to reveal the spatial distribution of the microorganisms involved in phenol degradation.One advantage of investigating a granular sludge is that there is a clear concentration gradient of phenol across the depth of the granules[17].Thus,the microorganisms that are present at different depths of the granules are exposed to different levels of phenol concentrations. In other words,the distribution and growth of microorganisms in the granules are defined by the concentration gradient of phenol and its byproducts across the depth of the granule. Furthermore, crossfeeding relationships can be observed by the in situ staining of specific microbial groups. In this study, for the first time, we explain the roles and interactions of the microorganisms found in phenol-degrading anaerobic granule consortia.
2. Materials and methods
2.1. Reactor operation and granular sludge sampling
A laboratory-scale UASB reactor (11 L in volume)was operated for more than 1000 d under mesophilic conditions(35°C)by feeding phenol as the sole carbon source,with supplementary nutrients(Appendix A Table S1). The hydraulic retention time (HRT) of the UASB reactor was set at 9.2 h. The UASB reactor was started up by feeding 630 mg·L-1of phenol, which is equivalent to 1500 mg of chemical oxygen demand(COD)determined by using potassium dichromate as an oxidant per liter(CODCr·L-1).The phenol concentration was gradually increased to 1260 mg·L-1(3000 mg CODCr·L-1), which corresponded to a COD loading of 7.8 kg CODCr·(m3·d)-1. The COD-removal efficiency of the reactor was in the range of 60%-80%.
For this study, granular sludge from the bottom of the UASB reactor was sampled. The sample was used for DNA extraction after being washed twice with phosphate-buffered saline (PBS).The sample used for FISH analysis was washed twice with PBS,fixed in 3% paraformaldehyde solution for 15 h at 4 °C, washed twice with PBS again, and then stored in ethanol/PBS solution at-20 °C.
2.2. DNA extraction, cloning, phylogenetic analysis, probe design, and validation
DNA extraction was carried out using ISOIL for Beads Beating kit(NIPPON GENE,Japan).Amplifications of 16S rRNA genes for bacteria and archaea were conducted using the primer pairs Eub8FUniv1500R [18] and Arc109F-Univ1500R [19], respectively. After treatment with a MinElute PCR Purification Kit(Qiagen,Germany),the polymerase chain reaction(PCR)products were cloned using a TOPO TA Cloning Kit (Invitrogen, USA), following the manufacturer’s instructions.A total of 117 bacterial clones and 38 archaeal clones were randomly picked. Approximately 600 bases were sequenced including the V3-V4 region of the 16S rRNA gene. The obtained sequences were clustered into operational taxonomic units(OTUs),with a threshold value of 97%sequence identity using the mothur algorithm [20]. For select important OTUs, the full length of the gene was sequenced. The sequences were deposited into the DNA Databank of Japan (DDBJ; numbers are shown in Appendix A Table S2).Phylogenetic analysis and probe design were carried out using ARB software with the SILVA Release 138 database [21]. The designed probes were evaluated using mathFISH[22]. In cases where the affinity was expected to be low, locked nucleic acid (LNA) was introduced [23]. Probe validation was carried out using clone-FISH, as described elsewhere [24].
2.3. In situ hybridization and confocal laser scanning microscopy
The oligonucleotide probes used in this study are listed in Table 1 [25-27]. A mixture of an equal amount of the Eub338 I&IV and Eub338 II&III probes was used to detect bacteria.Hybridization was performed at 46 °C for 3-6 h in a hybridization buffer (0.9 mol·L-1NaCl, 20 mmol·L-1Tris-HCl (pH 7.5), 0.01%sodium dodecyl sulfate) containing 0.5 μmol·L-1of each probe.The hybridization stringency was adjusted by adding formamide to the hybridization buffer (Table 1). Excess probes were removed by immersing the slides in the hybridization buffer with formamide for 15 min at 48 °C. To enhance the signal intensity of some probes, fluorescent dyes were labeled at both the 3′and 5′ends [28]. After drying, the samples were covered with ProLong Gold (Invitrogen) and evaluated using an epifluorescent microscope (BX50, Olympus, Japan) or a confocal laser scanning microscope (LSM710, Carl Zeiss Microscopy, Japan). Cell abundances(as percentages) were calculated by dividing the number of FISHpositive cells by the number of 4′,6-diamidino-2-phenylindole(DAPI)-stained cells.
3. Results and discussion
3.1. Microbial community composition
Bacterial and archaeal libraries were constructed by sequencing the 117 and 38 clones,respectively.Out of all the clones,30 bacterial and five archaeal OTUs were obtained. The bacterial community composition is shown in Table S2. Three OTUs were close to the known phenol-degrading bacteria, S. aromaticivorans (OTUs B1 and B15) and C. phenolicus (OTU B4). S. aromaticivorans is capable of degrading phenol to acetate in syntrophic association with methanogens in an anaerobic environment [11]. Likewise, C. phenolicus is known to convert phenol to benzoate [12].
The assignment of OTU B4 to C. phenolicus was not straightforward. Although the basic local alignment search tool (BLAST)search using the National Center for Biotechnology Information(NCBI) database showed a higher sequence identity of OTU B4 to
Pelotomaculum terephthalicicum (P. terephthalicicum, 96%) as compared with C. phenolicus (95%), direct sequence matching showedOTU B4 being closer to C. phenolicus (1431 bases match in 1499 bases) than to P. terephthalicicum (1422 bases match in 1488 bases; Table S2). In order to further clarify the phylogenetic position of OTU B4,a phylogenetic tree was constructed,which clearly indicated that the OTU was more closely related to the members of Cryptanaerobacter than to those of Pelotomaculum (Fig. 1(a)). Such an anomaly in the method of data analysis for taxonomic assignment could explain why many studies have mentioned the presence of Pelotomaculum rather than Cryptanaerobacter in phenol-degrading consortia [2,15]. Next-generation sequencing provides a higher number of sequence reads,albeit in short lengths that may not be sufficient to distinguish between Cryptanaerobacter and Pelotomaculum. Even after obtaining nearly the full length of the 16S rRNA gene sequence, the results could still be biased toward Pelotomaculum, especially if phylogenetic analysis is not conducted. Ju et al. [5] reported a high relative abundance of Cryptanaerobacter and the absence of Pelotomaculum relatives in ambient, mesophilic, and thermophilic anaerobic phenoldegrading consortia after phylogenetic analysis with full-length or nearly full-length 16S rRNA gene sequences. These results indicate that phylogenetic analysis with nearly the full length of the 16S rRNA gene sequence is necessary for more accurate taxonomic assignment of sequences belonging to Cryptanaerobacter.
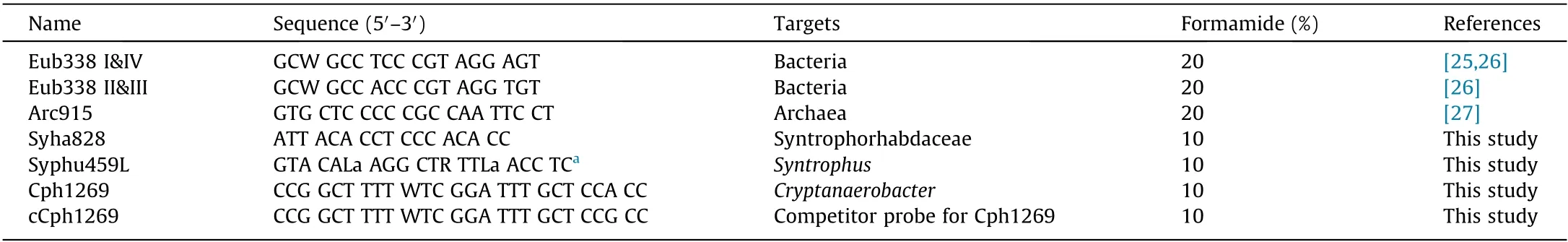
Table 1 List of probes used in the study.
The second most abundant OTU (OTU B2, 17% relative abundance in the library) was closely related to Syntrophus buswellii,which is known to convert benzoate to acetate[29].The third most abundant OTU (OTU B3, 10% relative abundance) belonged to the phylum FCPU426. The metagenome assembled genomes in this phylum were constructed using samples obtained from thawing permafrost [30], but any detailed report on their metabolic functions is not yet available.The OTUs belonging to Desulfovibrio were also obtained in high abundance: B6 (four clones), B9 (three clones),and B13(two clones).Several other studies have also mentioned the presence of Desulfovibrio in phenol-degrading consortia.Chen et al.[9]observed the presence of Desulfovibrio in a mesophilic phenol-degrading enrichment culture, but did not discuss its role.Desulfovibrio was likely to be involved in hydrogen consumption using sulfate (Table S1). Its contribution to hydrogen consumption may have resulted in a low retrieval of archaeal clones related to hydrogenotrophic methanogens (Table S2).
Regarding the archaeal library(Table S2),the OTUs belonging to Methanosaeta, an acetoclastic methanogen, were dominant because acetate is a major daughter product during anaerobic phenol degradation. Low relative abundances of OTUs were obtained,which were related to hydrogenotrophic methanogens—that is,Methanolinea and Methanobacterium.
3.2. Probe design and validation
The phylogenetic trees constructed for the OTUs close to Syntrophorhabdus, Cryptanaerobacter, and Syntrophus are presented in Fig. 1. Three oligonucleotide probes—namely, Cph1269, Syha828,and Syphu459L—were designed to detect Cryptanaerobacter,Syntrophorhabdaceae,and Syntrophus, respectively(Table 1). Phylogenetic coverages of the designed probes are shown in Fig.1.The designed probes were further validated and optimized using the clone-FISH method (Appendix A Fig. S1). Among the three probes,Syphu459,which targets Syntrophus,showed a low signal intensity with a DNA probe, even after labeling with a fluorophore on both the 5′and 3′ends [28]. Therefore, LNA was introduced to enhance the hybridization efficiency [23]. After substituting two adenine bases (Syphu459L), brighter signals were obtained. For the detection of Cryptanaerobacter, probe Cph1271 (Fig. S1) was initially designed. This probe had only one base mismatch at the 3′end to Pelotomaculum propionicum (P. propionicum). Therefore, a competitor probe, cCph1271 (Fig. S1), was also designed and applied in combination with Cph1271. Even with cCPh1271, non-specific hybridization with P. propionicum could not be eliminated. The probe was then elongated by adding two cytosine bases at the 3′end(named Cph1269)and was applied with the competitor probe,cCph1269. This combination resulted in a clear distinction between Cryptanaerobacter and P. propionicum.
3.3. In situ localization of phenol-degrading microorganisms in UASB granular sludge
The designed probes were applied to the granule samples after ultrasonic homogenization. All three microbial groups—that is,Syntrophorhabdaceae, Cryptanaerobacter, and Syntrophus—were successfully detected(Fig.2).The percentages of the cells detected by each probe were 22% ± 0.6% for Syntrophorhabdaceae, 3.5% ±1.5% for Cryptanaerobacter, and 14.4% ± 0.6% for Syntrophus. This result is in good agreement with their relative populations as found in the clone library.
Next,thin slices of the granule samples were prepared to reveal the distribution of phenol-degrading consortia along the depth of the granule. Initially, the probes targeting bacteria and archaea were applied.The results indicated that bacteria and archaea were distributed throughout the granules. In the center of the granule,there was an inactive zone or mineral-like material emitting autofluorescence(Fig.3).Next,the three designed probes were applied,targeting Syntrophorhabdaceae,Cryptanaerobacter, and Syntrophus simultaneously. It was found that Cryptanaerobacter and Syntrophus were closely spatially associated and were located on the outer part of the granule,while Syntrophorhabdaceae was present in the deeper part of the granule (i.e., between the layer of Cryptanaerobacter and Syntrophus and the central inactive zone;Fig. 3).
3.4. Ecological perspective on the anaerobic phenol-degrading community
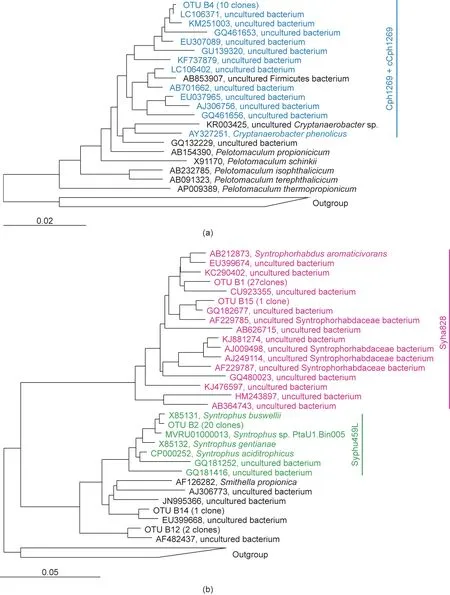
Fig.1. Phylogenetic positions of OTUs related to(a)Cryptanaerobacter and(b)Syntrophorhabdus and Syntrophus. The numbers of clones obtained are shown in parentheses.Coverages of the designed probes are shown in colors:blue for Cph1269,pink for Syha828,and green for Syphu459L.The 16S rRNA gene-based tree was constructed using the neighbor-joining method implemented in the ARB program. Scale bars indicate the number of nucleotide changes per sequence position.
In this study,we successfully demonstrated the in situ distribution of phenol-degrading microorganisms in granular sludge used to treat phenol-containing wastewater. Random distribution of bacteria and archaea in phenol-degrading granules and biofilms has been reported previously[7,9,10,14],and our results were consistent with those observations. Interestingly, in this study, FISH analysis showed a clear distinction in the distribution of different bacterial species in the granular sludge.The specific microbial distribution in the granule likely occurred due to factors such as phenol degradability, metabolic association, resistance to phenol toxicity, and difference in the growth rates of Cryptanaerobacter and Syntrophorhabdaceae. Cryptanaerobacter may have a faster growth rate than Syntrophorhabdaceae, which helped the former obtain an ecological niche on the outer part of the granules by outcompeting Syntrophorhabdaceae for phenol utilization. Benzoate, produced by Cryptanaerobacter, can be utilized by Syntrophorhabdus and Syntrophus, according to the information obtained from the clone library(Table S2).The FISH results showed a high abundance of Syntrophus spatially close to Cryptanaerobacter,indicating that Syntrophus outcompetes Syntrophorhabdaceae for benzoate utilization.Dominance of Syntrophus over Syntrophorhabdaceae in benzoate utilization has been reported previously[9];in the same work, a population shift from Syntrophorhabdaceae to Syntrophus was observed when the substrate was switched from phenol to benzoate.These observations suggest that phenol degradation takes place via syntrophic association between Cryptanaerobacter and Syntrophus on the outer part of the granules.

Fig. 2. Photomicrographs of phenol-treating granular sludge samples after in situ hybridization with (a)the Syha828 probe (Syntrophorhabdaceae), (b) the Cph1269 probe (Cryptanaerobacter), and (c) the Syphu459L probe (Syntrophus). Each double panel depicts DAPI (left) and probe staining (right). The bar represents 10 μm.
Although the members of Syntrophorhabdaceae have slower growth rates, they may have a higher affinity to phenol compared with Cryptanaerobacter and have found their ecological niche in the deeper part of the granules.The reactor in this study was fed with a high concentration of phenol(1260 mg·L-1),but the formation of a steep phenol concentration gradient might occur along the granule depth as a result of microbial phenol degradation [17]. Therefore,the phenol concentration in the deeper part of the granules could be significantly low, which could have facilitated the survival,growth, and prevalence of Syntrophorhabdaceae deeper within the granules. Chen et al. [14] reported the presence of Syntrophorhabdaceae on the surface of biofilms developed on carbon-activated granules in a full-scale anaerobic fluidized bed reactor, when fed with low concentrations of phenol (25-30 mg·L-1). In addition, Syntrophorhabdaceae was found to be the dominant microorganism in an enrichment culture with low concentrations of phenol [31], but Pelotomaculum and Syntrophus were found to be dominant when the phenol concentrations were high [2,7,10]. Although no studies have focused on the affinity of these microorganisms for phenol, the microbial distribution and possible phenol concentration gradient in the granules in this study suggest that Syntrophorhabdaceae could have a higher affinity for phenol compared with Cryptanaerobacter.
The niche differentiation shown in this study also suggests various roles for the anaerobic organisms in phenol degradation.Some studies have reported that Syntrophorhabdus is likely to be the key microorganism in degrading phenol to benzoate, after which benzoate degradation is carried out by Syntrophus and Pelotomaculum[4,14].However,it is less likely that Syntrophorhabdus always produces benzoate as an intermediate to feed other microorganisms because there is little benefit for Syntrophorhabdus in doing so[16]. The FISH results showed that Syntrophorhabdaceae existed by itself in the deeper part of the granules, rather than together with Syntrophus. This finding indicates that Syntrophorhabdaceae is not in a cross-feeding relationship with Syntrophus in the granules. On the other hand,there are reports of random distribution of Syntrophorhabdaceae and Syntrophus near the surface of a biofilm treating low-strength phenol [14], which suggest different microbial interactions than our findings. It is possible that the microbial interactions are affected by several different environmental factors, such as phenol concentration, organic loading,and biofilm thickness.
In this study,we demonstrated the importance of phylogenetic analysis performed with nearly the full length of the 16S rRNA gene sequence for the correct identification of Cryptanaerobacter,in order to distinguish it from Pelotomaculum. It is possible that some of the previous studies may have misidentified Cryptanaerobacter as Pelotomaculum in phenol-degrading consortia. This study identified the microorganisms responsible for phenol degradation and suggested their roles in an anaerobic granular biofilm environment.Finally,this study was able to answer the two key questions:①Syntrophorhabdaceae obtains energy by degrading phenol to acetate,and does not provide benzoate to neighboring microorganisms; and ②Syntrophus obtains benzoate as a primary substrate from Cryptanaerobacter.
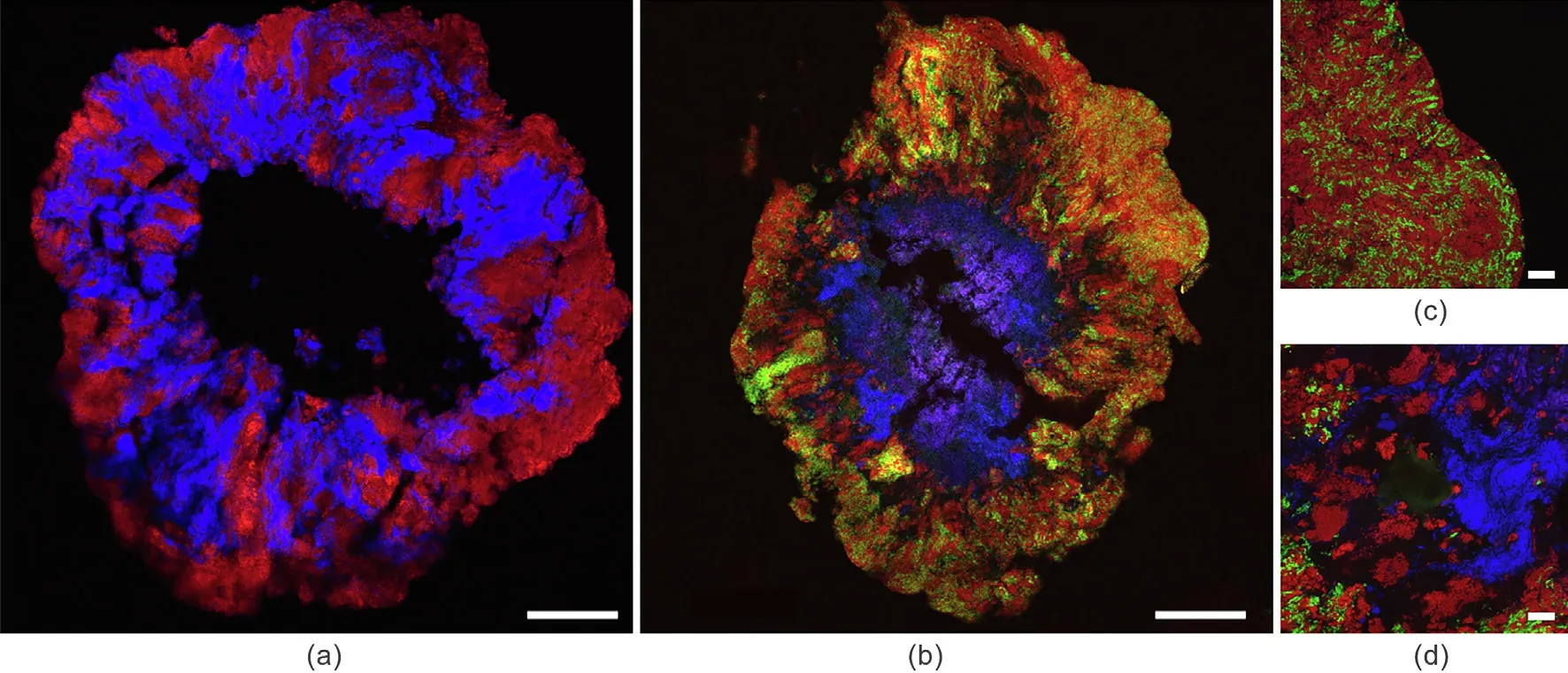
Fig. 3. In situ hybridization of sections of the phenol-treating granules. The sections were simultaneously hybridized with (a) Eub338 mix for bacteria (red) and Arc915 for archaea (blue),or (b) with the Cryptanaerobacter-targeting Cph1269 probe (green),the Syntrophu-targeting Syphu459L probe (red),and the Syntrophorhabdaceae-targeting Syha828 probe (blue). Higher magnification views are shown of (c) outer and (d) deeper parts of the cross-section simultaneously hybridized with three probes (Cph1269,Syphu459L, and Syha828). Bars represent (a, b) 200 μm and (c, d) 20 μm.
Acknowledgments
We thank Dr. Madan Tandukar of Höganäs Environment Solutions, LLC (USA) for reading the manuscript. This research was supported by Grant-in-Aids for Scientific Research (B)(JP18H01564) from the Japan Society for the Promotion of Science.
Compliance with ethics guidelines
Kengo Kubota, Kei Igarashi, Masayoshi Yamada, Yasuyuki Takemura, Yu-You Li, and Hideki Harada declare that they have no conflict of interest or financial conflicts to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.05.012.

- Engineering的其它文章
- Science and Technology for Combating Global Water Challenges
- Brain-Computer Interface Speaks Up
- Solar Geoengineering to Reduce Global Warming—The Outlook Remains Cloudy
- Pandemic Scrambles the Semiconductor Supply Chain
- A Multi-Stage Green Barrier Strategy for the Control of Global SARS-CoV-2 Transmission via Cold Chain Goods
- Next-Generation Imaging: New Insights from Multicolor Microscopy in Liver Biology and Disease