
高通量测序法对糖尿病患者及健康人群粪便微生物的群落分析
2022-04-17 18:40王梦姣杨国鹏郑天骄李青山
右江医学 2022年3期
王梦姣 杨国鹏 郑天骄 李青山
【摘要】 目的 通過寻找出2型糖尿病患者的粪便指示微生物,为后续进一步开发潜在糖尿病患者试剂盒做准备,并探讨母系亲缘关系与人群粪便微生物多样性的相关性。
方法 从粪便微生物入手,利用高通量测序法对健康人群及糖尿病患者进行粪便微生物群落的分析和比较。同时,从多样性方面对具有母系遗传关系的四代人类粪便微生物进行比较。
结果 与健康人群相比,糖尿病人群粪便微生物多样性显著弱于健康人群;厚壁菌门(Firmicutes)、梭菌纲(Clostridia)、梭菌目(Clostridiales),Ruminococcaceae_UCG-013和粪杆菌属(Faecalibacterium)是2型糖尿病患者粪便指示微生物。三相图结果表明,只有布劳特氏菌属(Blautia)连续出现在四代人类粪便微生物群落中;PCoA及LEfSe的差异分析结果表明,四代人粪便微生物群落相似性较差。
结论 厚壁菌门、梭状芽孢杆菌纲、梭状芽孢杆菌目、Ruminococcaceae_UCG-013和粪杆菌属可作为筛选潜在糖尿病人群的粪便指示微生物。粪便微生物群落与母系遗传关系相关性不强。
【关键词】 粪便微生物;2型糖尿病;指示微生物;母系亲缘关系;相关性
中图分类号:R587.1 文献标志码:A DOI:10.3969/j.issn.1003-1383.2022.03.002
Analysis of fecal microbial community of diabetic patients and healthy people by high-throughput sequencing
WANG Mengjiao, YANG Guopeng, ZHENG Tianjiao, LI Qingshan
(School of Biological Science and Engineering, Shaanxi University of Technology, Hanzhong 723000, Shaanxi, China)
【Abstract】 Objective To prepare for further development of potential diabetic kits through finding out fecal indicator microorganisms of T2DM patients, and to explore the correlation of maternal relationship and fecal microbial diversity.
Methods Starting with fecal microorganism, high-throughput sequencing was used to analyze and compare fecal microbial communities in healthy people and T2DM patients. At the same time, four generations of human fecal microorganisms with maternal genetic relationship were compared in terms of diversity.
Results Compared with healthy population, the diversity of fecal microorganism in T2DM patients was significantly weaker than that in healthy population. Firmicutes, Clostridia, Clostridiales, Ruminococcaceae_UCG-013 and Faecalibacterium were indicators of fecal microorganisms in T2DM patients. The results of three-phase diagram showed that only Blautia appeared continuously in the four generations of the community of human fecal microorganisms. The difference analysis results of PCoA and LEfSe showed that the similarity of fecal microorganisms communities of the four generations was poor.
Conclusion Firmicutes, Clostridia, Clostridiales, Ruminococcaceae_UCG-013 and Faecalibacterium can be used as screening indicators for fecal microorganisms among potential T2DM patients, and correlation between fecal microorganisms community and maternal genetic relationship is not strong.
【Key words】 fecal microorganism; T2DM; indicator microorganism; matrilineal relationship; correlation
糖尿病是一种以高血糖为特征的代谢性疾病,按发病机制分为1型糖尿病(type 1 diabetes mellitus, T1DM)和2型糖尿病(T2DM);2型糖尿病约占糖尿病病例的90%,是一种由胰岛素抵抗和细胞缺陷引起的胰岛素敏感性受损导致的慢性代谢紊乱[1]。虽然目前我们已经对T2DM发生、发展及相关的生理生化知识有了很多了解,也发展出较多的2型糖尿病治疗方法。但是随着近年来全球2型糖尿病患者数量的过快增长,除了现有的2型糖尿病防治措施以外,我们依然需要进一步了解和探索2型糖尿病的发病机理和相关生理生化指标,从中寻找能够甄别潜在糖尿病患者的方法,将2型糖尿病从治疗推向预防的阶段。
当前许多研究结果都表明,2型糖尿病患者的肠道菌群与健康成年人显著不同。有研究结果揭示,与健康人群肠道微生物相比,糖尿病患者的厚壁菌门(Firmicutes)和类梭状芽孢杆菌纲(Clostridia)的比例显著降低;厚壁菌门、拟杆菌门(Bacteroidetes)与受试者血糖呈显著正相关关系。另一个研究小组利用宏基因组关联技术对345名受试人员进行了肠道微生物测序,其研究结果表明2型糖尿病患者具有一定程度的肠道微生物生态失衡,丁酸盐产生菌数量和种类大量下降,各种病原微生物种类和数量有所增加。以上研究结果均表明,2型糖尿病与人体肠道微生物菌群存在一定相关性。
人体肠道微生物的建立及稳固主要取决于个体出生方式、遗传因素和居住环境(饮食习惯)三者的协同作用。人体肠道微生物最初是由其出生方式及母体的基因型决定;如果婴儿出生方式是顺产,其肠道微生物主要来源于母体生殖道及粪便微生物,如果婴儿是剖腹产方式出生,其肠道微生物的多样性更偏向于母体皮肤部位的微生物。待婴儿成长至3岁以后,肠道微生物则逐渐趋于稳定,此时肠道微生物构成则主要取决于遗传因素、环境因素及饮食习惯。
2型糖尿病作为一个非常复杂的代谢性疾病,很多研究指出其与遗传因素具有较强的相关性。尼尔森的研究表明,环境因素和遗传因素共同促成了2型糖尿病的发病机制。人体全基因组关联性分析结果也确定了大量在基因型上与2型糖尿病相关的变异情况;BARBITOFF和他的团队已经选定出部分单核苷酸多态性(single nucleotide polymorphism,SNP)位点作为2型糖尿病的遗传标记。
本研究从人体粪便微生物入手,利用高通量测序法,以健康人群为对照,对糖尿病患者粪便微生物进行了测序分析和比较,拟尋找出2型糖尿病患者的粪便指示微生物,为后续进一步开发潜在糖尿病患者试剂盒做准备;同时,本研究还从多样性方面对具有母系遗传关系的四代人类粪便微生物进行了比较,探讨母系亲缘关系是否与人群粪便微生物多样性存在一定相关性。
1 材料与方法
1.1 研究对象和取样方法
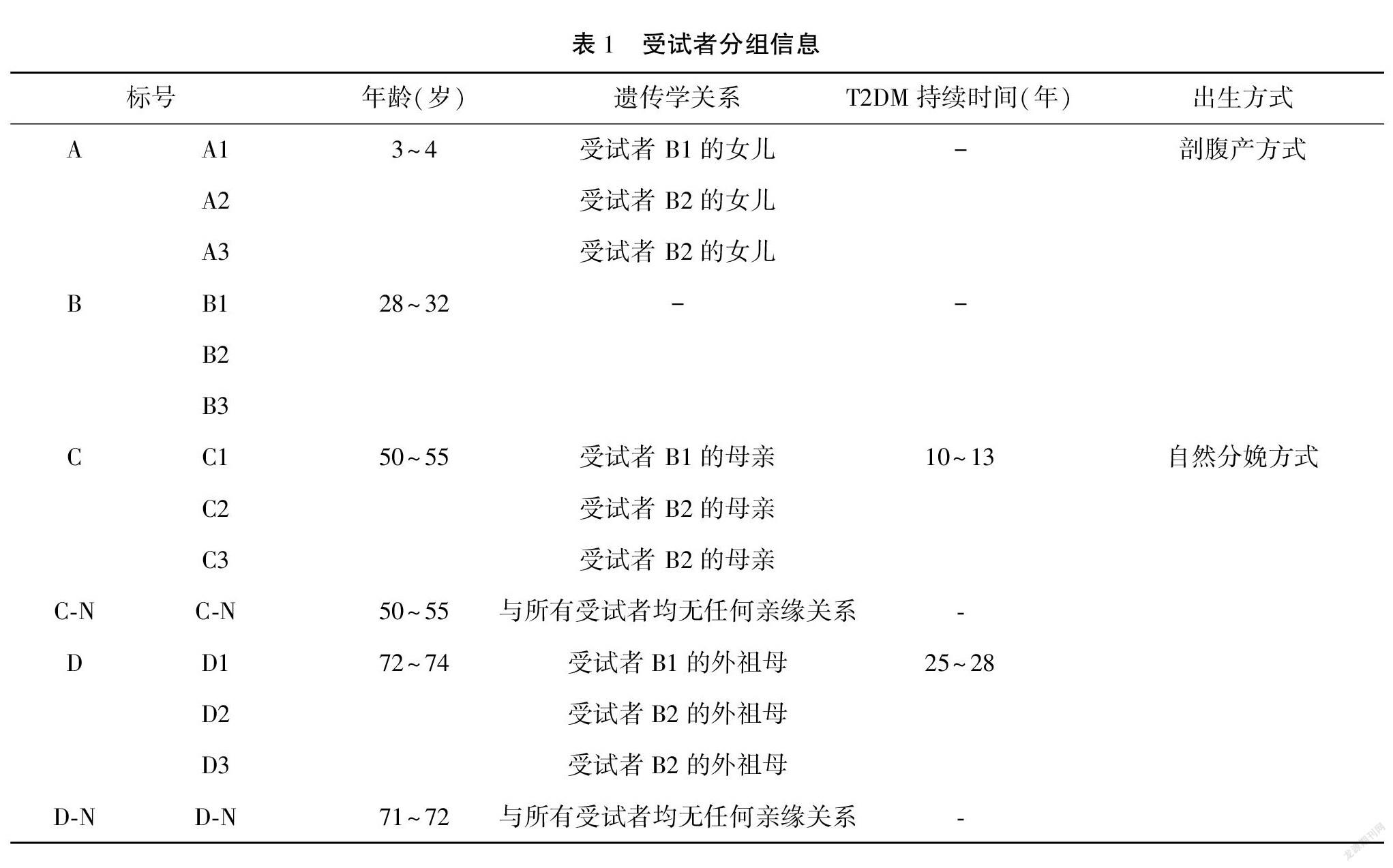
受试对象共18人,其中每3人分为一组,分别为A、B、C、D、C-N和D-N组,其中,A、B、C和D组具有母系遗传亲缘关系,C-N、D-N组与其他四组受试者没有任何亲缘关系,为对照组,均为健康人群。以上所有受试对象除糖尿病组以外,其余健康状况良好,均无任何其他遗传疾病。C组糖尿病治疗方法为在早饭和晚饭前30分钟,注射胰岛素Gansulin R(通化东宝制药有限公司,中国),D组糖尿病治疗方法为在早饭和晚饭前30分钟,注射胰岛素Novolin R (诺和诺德公司,丹麦)。具体分组信息见表1。所有粪便样本均由受试者自行收集后,并迅速冻存至-80℃条件下。
1.2 粪便DNA提取及测序
用DNA提取试剂盒(SPINeasy DNA Kit for Feces,MP生物试剂公司,圣安娜,CA,USA)进行粪便样品基因组DNA提取。用引物806R和515F扩增细菌16S rRNA基因的V4区,PCR反应体系为50 μL,由25 μL 2x Premix Taq (Takara Biotechnology, 大连,中国)、1 μL浓度为10 mM的引物806R、1 μL浓度为10 mM的引物515F、3 μL of DNA(20 ng/μL)模板以及21 μL of dd H2O。扩增程序为94℃ 5 min,at for initialization,94℃ 30 s,52℃ 30 s,72℃ 30 s,30个循环,72℃10 min。
扩增结束之后,用1%琼脂糖浓度的电泳来检测扩增片段的长度及产物浓度。用NEBNext Ultra DNA建库试剂盒将扩增长度在290~310 bp的DNA片段进行DNA扩增子文库的构建,待构建好文库后,用Illumina Hiseq2500平台对构建的扩增子文库进行PE250测序(Guangdong Magigene Biotechnology Co.,Ltd. Guangzhou, China)。文库OTU(operational taxonomic unit)数据的获取及测序过程以及OTU序列注释过程参考Schwiertz A的实验方法。
1.3 数据统计分析
试验中所有数据结果均为三个平行样品的平均值,并对结果进行了方差分析。分析不同样品(组)之间共有、特有的OTU数,并利用R软件绘制维恩图。用Chao1指数、香农指数、辛普森指数来进行样品Alpha多样性分析。使用QIIME(V1.9.1)软件进行分析,用R(V2.15.3)软件计算出结果。其中,用Chao1指数表征样品群落丰富度,用香农指数和辛普森指数表征样品群落多样性。三个指数计算公式见下:
Chao1=Sobs+F21/2F2 (Chao, 1984) ;Shannon=-∑si=1pilog2pi(Colwell, and Coddington, 1994) ; Simpson= 1-∑p2 i (Adrain, 2000)。
选取总体相对丰度排在前50且有属分类信息的OTU代表性序列,构建系统发育树,系统发育树构建参考SAITOU等的文献。分别选取A、B和C受试者粪便样品中各分类级别中平均丰度排名前10位的优势门、优势纲、优势目、优势科及优势属,利用R软件绘制三元相图。利用相同选择方法,使用R软件绘制受试者B、C和D受试者粪便样品的三元相图。基于Weighted Unifrac和Unweighted Unifrac距离矩阵,使用qiime2和ggplot2软件包进行Principal co-ordinates analysis(PCoA)分析并绘图。使用LEfse软件进行组间物种的差异显著分析:首先使用non-parametric factorial Kruskal-Wallis(KW)sum-rank test(非参数因子克鲁斯卡尔-沃利斯秩和检验)检测不同分组间丰度差异显著的物种,然后用成组的Wilcoxon秩和检验来进行两两组间差异性判断,最后用线性判别分析(LDA)来实现降维和评估差异显著物种的影响大小(即为LDA Score),默认设置LDA Score的筛选值为2,结果即为各组内的biomarker。
2 结 果
2.1 糖尿病患者与健康人群粪便微生物群落结构分析
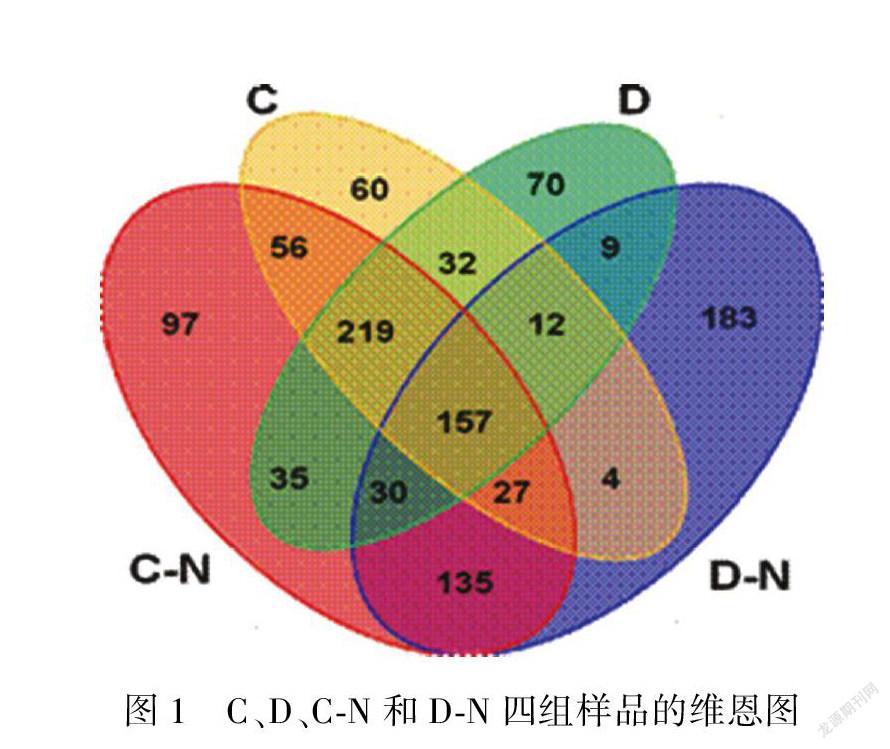
为分析健康人群与糖尿病患者粪便样品的群落结构,本研究首先通过分析不同样品之间共有和特有的OTU数,绘制了C组、D组、C-N组和D-N组的维恩图(图1)。从图1中可以看出,四组粪便样品共检出1126种微生物,其中D-N组样品的特有微生物种类数量最多,为183种;C组样品的特有微生物种类数量最少,为60种;有157种微生物出现在四组样品中,为四组样品的共有微生物。
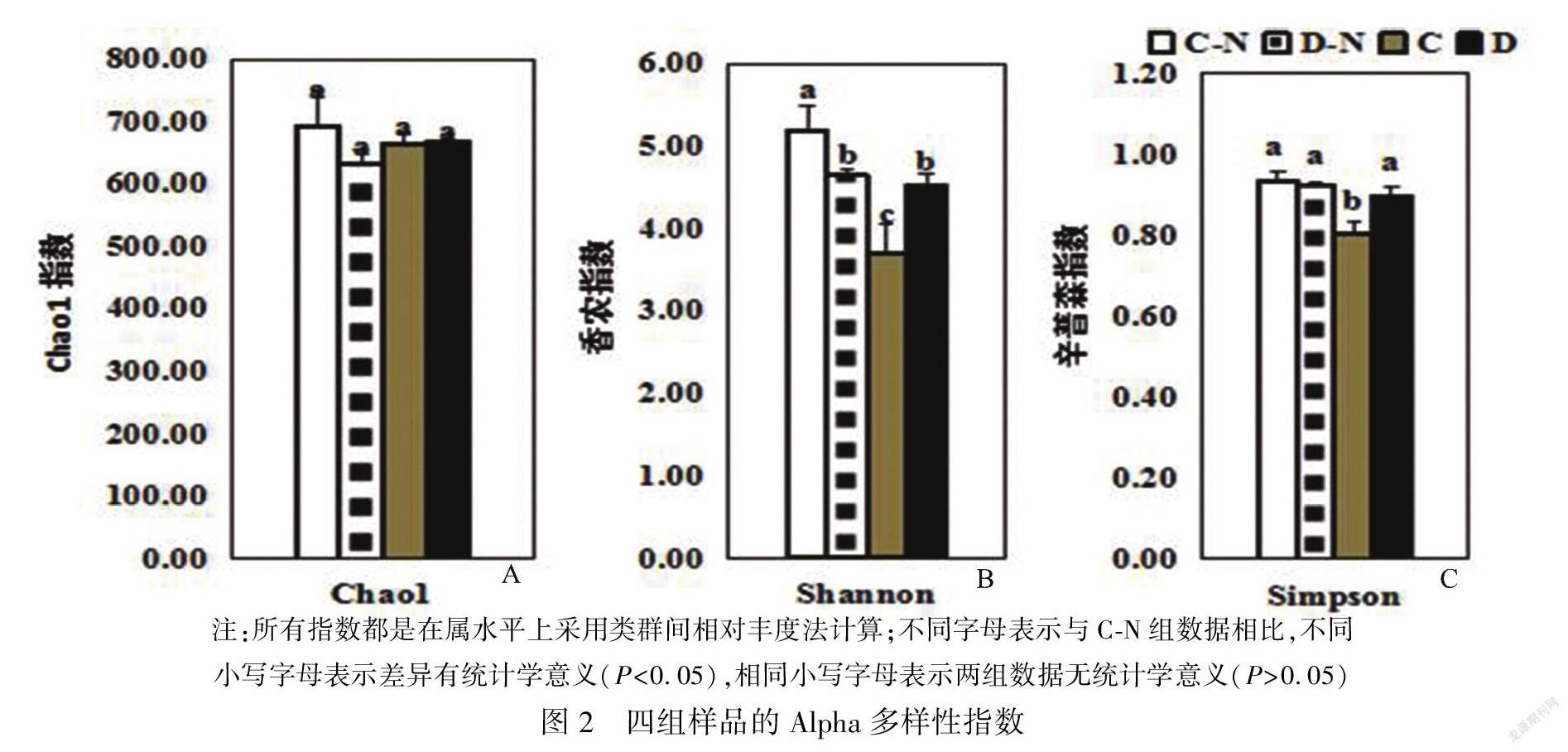
为了明确健康人群与糖尿病人群在微生物分布多样性方面的差异,本研究对以上四组的多样性指数(Chao1指数、香农指数和辛普森指数)进行了数据分析。从图2中可以看出,样品C-N组的微生物物种丰富度最高,D-N组丰富度最低(图2A);样品C-N组的物种多样性也最高(图2B、C)。
为进一步从不同分类水平上对健康人群及糖尿病群体微生物群落的差异性进行分析,本研究分别从不同分类水平对四组样品微生物群落进行了分类比较(图3)。四组样品中分离的微生物分布在31个门,其中厚壁菌门、拟杆菌门、变形菌门(Proteobacteria)、放线菌门(Actinobacteria)和疣微菌门(Verrucomicrobia)為优势菌门,占整个微生物分布的99%以上(图3A)。和健康人群粪便微生物样品(C-N、D-N)相比,C组样品变形菌门微生物分布显著增加,厚壁菌门微生物分布显著下降;D组样品疣微菌门微生物分布显著增加,厚壁菌门微生物分布显著下降(图3A)。即在门的分布水平上,健康人群与糖尿病患者粪便微生物的差异主要集中在变形菌门、厚壁菌门及疣微菌门这三个门的分布上。放线菌纲(Actinobacteria)、红蝽菌纲(Coriobacteriia)、梭菌纲(Clostridia)、杆菌纲(Bacilli)、δ变形菌纲(Deltaproteobacteria)、γ-变形菌纲(Gammaproteobacteria)和疣微菌纲(Verrucomicrobiae)是四组样品从纲的水平上进行分类所获得的优势菌纲(图3B)。厚壁菌门下的梭菌纲在健康人群的粪便微生物中分布显著多于糖尿病患者样品,C组样品中γ-变形菌纲显著多于健康人群样品,D组样品则是疣微菌纲显著多于健康人群样品(图3B)。从图3C中可以看出,四组样品中相对丰度大于1%微生物分布在11个目中,与健康人群粪便样品相比,C组样品的梭菌目(Clostridiales)的相对丰度显著降低,肠杆菌目(Enterobacteriales)和假单胞菌目(Pseudomonadales)相对丰度则显著升高;D组样品的梭菌目(Clostridiales)的相对丰度相对于健康人群也显著降低,假单胞菌目(Pseudomonadales)和疣微菌目(Verrucomicrobiales)相对丰度则有所上升(图3C)。从图3D中看出,与健康人群相比,糖尿病患者粪便微生物中链球菌科的相对分布略有下降,而理研菌科(Rikenellaceae)、韦荣氏菌科(Veillonellaceae)、肠杆菌科(Enterobacteriaceae)、莫拉氏菌科(Moraxellaceae)以及疣微菌科(Akkermansiaceae)的相对丰度则有所上升。
在四组样品中,所有微生物分布在584个属种,其中有26个属的相对丰度大于1%,这些属分别是双歧杆菌属(Bifidobacterium)、拟杆菌属(Bacteroides)、另枝菌属(Alistipes)、狄氏副拟杆菌属(Parabacteroides)、乳酸菌属(Lactobacillus)、魏丝氏菌属(Weissella)、链球菌属(Streptococcus)、Agathobacter、布劳特氏菌属(Blautia)、Fusicatenibacter、罗斯氏菌属(Roseburia)、Intestinibacter、Romboutsia、粪杆菌属(Faecalibacterium)、Ruminococcaceae_UCG-013、Ruminococcus1、Ruminococcus2、Subdoligranulum、Coprostanoligenes_group、Erysipelotrichaceae_UCG-003、苏黎世杆菌属(Turicibacter)、埃希氏杆菌属(Escherichia/Shigella)、克雷白氏杆菌属(Klebsiella)(图3E)。与健康人群相比,糖尿病患者粪便样品中链球菌属、粪杆菌属和Subdoligranulum的相对分布显著降低,另枝菌属、乳酸菌属、Coprostanoligenes_group和埃希氏杆菌属的相对丰度显著上升(图3F)。
2.2 母系遗传关系与粪便微生物群落的相关性分析
本研究首先利用三相图探讨母系遗传关系与粪便微生物群落的相关性;在三元相图中,三个顶点粪便代表三个样本,圆形图案代表三元相图中的物种,圆的大小与相对丰度成正比。通过分析圆形与三个顶点之间的距离可以分析出,不同样本的相近核心微生物群落种类。从受试B、C和D组的粪便微生物的三元相图中可以看出,在门的分类水平上,酸杆菌门(Acidobateria)位于三元相图的中间,即该菌门在三组样品中具有相似的丰度(图4A);在属的分类水平上,布劳特氏菌属在三组样品中也具有相似的丰度(图4E)。然而,在其他分类水平上,均没有找到具有相似丰度的微生物菌种(图4B、图4C及图4D)。
在受试A、B和C组的三元相图中可以看出,厚壁菌门、放线菌门和绿弯菌门(Chloroflexi)在三组样品中具有相似的丰度(图5A);放线菌纲和Chlostridia,Bifidobacterialesc和梭菌目,疣微菌科、毛螺菌科、双岐菌科和Peptostreptococcacceae,双歧杆菌属和布劳特氏菌属分别在纲、目、科和属的水平上,在三组样品中具有相似丰度(图5B、C、D和E)。
3 讨 论
3.1 糖尿病患者与健康人群粪便微生物群落结构分析
两组糖尿病患者粪便样品的共有OTU数为420,是其他任意两组共有OTU数量最高的数值(图1);健康人群特异性OTU数分别为97和183(图1)。以上结果均表明,糖尿病患者的肠道菌群OTU数量相似且保守性较高,其特异性要显著低于健康人群。这一结果与前人在研究阿卡波糖对糖尿病早期患者肠道菌群影响中的部分结果一致。
在粪便微生物多样性指数分析方面,样品C-N组被检出的微生物数量最多,从多样性方面来看,健康人群粪便微生物多样性指数略高于糖尿病患者粪便微生物多样性指数(图2)。以上结果说明,糖尿病患者粪便微生物多样性相对较差。在前人研究粪便微生物分子特征的过程中,也发现了类似的结果。
通过在不同分类水平对健康人群与糖尿病人群粪便微生物群落差异性的分析发现,厚壁菌门、梭状芽孢杆菌纲、梭状芽孢杆菌目、Ruminococcaceae_UCG-013以及粪杆菌属在糖尿病患者粪便中相对丰度显著低于健康人群粪便样品(图3),这一结论与ARUMUGAM及SATO等学者的研究结果一致。本研究还发现,受试者C组的粪便微生物中,变形菌门、γ-变形菌纲、肠杆菌科、肠杆菌目、埃希氏肠杆菌属的相对丰度要显著高于其他所有受试组样品(图3);疣微菌门、疣微菌纲、疣微菌目、Akkermansi则在受试者D组粪便微生物中呈现较高的相对丰度。以上这两种微生物显著变化的情况也出现在HARTSTRA关于瑞典绝经后肥胖白人女性胰岛素抵抗发展预测的研究及代谢综合征患者的受供者粪便对胰岛素敏感性的相关研究中。出现这种在不同糖尿病患者粪便样品中不同微生物群落相对丰富显著不同的情况,可能是由于其患糖尿病时间不一致,采取治疗的药物也不完全一致,其代谢系统的调节方式也不完全一致造成的。
通过对以上结果的总结和分析,本研究认为,厚壁菌门、梭状芽孢杆菌纲、梭状芽孢杆菌目、Ruminococcaceae_UCG-013以及粪杆菌属为糖尿病患者粪便微生物的指示微生物。值得注意的是,目前的研究还不清楚造成这些微生物显著变化的原因是否与糖尿病患者肠胃蠕动变化相关,还是这些粪便微生物的变化直接引起血糖代谢问题。但是可以确定的是,这些发生群落相对丰度变化的微生物是可以用来作为诊断潜在糖尿病患者的重要微生物指標的。本研究的这一结果能够从微生物角度加强潜在糖尿病患者的可预测性及其微生物群落致病性改变的可预测性。
3.2 母系遗传关系与粪便微生物群落的相关性分析
本研究在探讨糖尿病患者与健康人群粪便微生物群落结构分析中就已经发现,虽然受试者C组和受试者D组均患2型糖尿病、存在母系遗传关系,但是其粪便微生物群落的相对丰度一致性不高,甚至还出现了不一致的情况。三元相图的结果表明,酸杆菌门和布劳特氏菌属在受试B、C和D组的粪便微生物中具有相似的丰度(图4);厚壁菌门、放线菌门、绿弯菌门,放线菌纲和Chlostridia,Bifidobacterialesc和梭菌目,疣微菌科、毛螺菌科、双岐菌科和Peptostreptococcacceae,双歧杆菌属和布劳特氏菌属分别在纲、目、科和属的水平上,在受试A、B和C组三组样品中具有相似丰度(图5)。综合以上结果发现,在所有A、B、C和D受试组样品中,只有布劳特氏菌属在四代具有相似丰度。
为了进一步分析样本之间的关系,本研究对粪便样本中的细菌基因组DNA进行PCoA分析。PCoA分析主要是通过一系列典型值和特征向量的排序,从多维数据中提取最重要的元素和结构。PCoA分析结果表明,同一组的不同样本聚类在一起,但是不同组之间样本距离相对较远(图6)。本研究还分别在门、纲、目、科和属的水平上,通过计算样本之间相对丰度差异程度的LEfSe得分来评估不同组粪便微生物组成的差异。LEfSe分析结果表明,四组样本微生物相对丰度的标志性微生物的相对丰度及数量均差异显著(图7)。
以上结果均表明,受试者粪便微生物群落多样性受遗传关系的影响较小。这一结论否定与本研究初期分析母系遗传关系和粪便微生物群落相关性分析的目标。这可能是由于人体肠道微生物群落受到遗传、年龄、性别和饮食等因素综合影响,单一利用粪便微生物群落分析,或单一利用高通量测序方法都不能直观地表述出二者的相互关系。
综上所述,本研究采用高通量测序法分析健康人群与糖尿病人群粪便微生物的差异性,并寻找母系遗传关系与糖尿病患者粪便微生物的相关性。研究结果表明,厚壁菌门、梭状芽孢杆菌纲、梭状芽孢杆菌目、Ruminococcaceae_UCG-013和粪杆菌属可作为筛选潜在糖尿病人群的粪便指示微生物。根据三元相图、PCoA分析及LEfSe分析结果,粪便微生物群落与母系遗传关系相关性不强。本研究结果对筛选潜在糖尿病患者及开发筛选潜在糖尿病患者试剂盒具有一定实验支持作用。
參 考 文 献
[1]KARAA A, GOLDSTEIN A. The spectrum of clinical presentation,diagnosis,and management of mitochondrial forms of diabetes.Pediatr Diabetes,2015,16(1):1-9.
[2]ZHANG F H, WANG M, YANG J J, et al. Response of gut microbiota in type 2 diabetes to hypoglycemic agents.Endocrine,2019,66(3):485-493.
[3]卢朝秀,吴飞飞.肠道微生物群与2型糖尿病的研究进展.中国微生态学杂志,2019,31(7):866-869.
[4]FORSLUND K, HILDEBRAND F, NIELSEN T, et al.Disentangling the effects of type 2 diabetes and metformin on the human gut microbiota.Nature, 2015, 528(7581): 262-266.
[5]QIN J J, LI Y R, CAI Z M, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes.Nature,2012,490(7418):55-60.
[6]BCKHED F, ROSWALL J, PENG Y Q, et al. Dynamics and stabilization of the human gut microbiome during the first year of life.Cell Host Microbe,2015,17(5):690-703.
[7]TANAKA M, NAKAYAMA J. Development of the gut microbiota in infancy and its impact on health in later life.Allergol Int,2017,66(4):515-522.
[8]CHEN Q H, LI M W, WANG X. Enzymology properties of two different xylanases and their impacts on growth performance and intestinal microflora of weaned piglets.Anim Nutr,2016,2(1):18-23.
[9]DECKER E, HORNEF M, STOCKINGER S.Cesarean delivery is associated with celiac disease but not inflammatory bowel disease in children.Gut Microbes,2011,2(2):91-98.
[10]JOST T, LACROIX C, BRAEGGER C P, et al. Vertical mother-neonate transfer of maternal gut bacteria via breastfeeding.Environ Microbiol,2014,16(9):2891-2904.
[11]NILSSON E, LING C.DNA methylation links genetics,fetal environment,and an unhealthy lifestyle to the development of type 2 diabetes.Clin Epigenetics,2017,9:105.
SHALIMOVA A, FADIEIENKO G, KOLESNIKOVA O,et al.The role of genetic polymorphism in the formation of arterial hypertension,type 2 diabetes and their comorbidity.Curr Pharm Des,2019,25(3):218-227.
BARBITOFF Y A, SEREBRYAKOVA E A, NASYKHO-VA Y A, et al.Identification of novel candidate markers of type 2 diabetes and obesity in Russia by exome sequencing with a limited sample size.Genes,2018,9(8):415.
[14]SCHWIERTZ A, TARAS D, SCHFER K, et al.Microbiota and SCFA in lean and overweight healthy subjects.Obesity:Silver Spring,2010,18(1):190-195.
[15]CAUSEY B D. Parametric estimation of the number of classes in a population.J Appl Stat,2002,29(6):925-934.
[16]COLWELL R K, CODDINGTON J A.Estimating terrestrial biodiversity through extrapolation.Philos Trans R Soc Lond B Biol Sci,1994,345(1311):101-118.
[17]ADRAIN J M, WESTROP S R, CHATTERTON B D E, et al.Silurian trilobite alpha diversity and the end-ordovician mass extinction.Paleobiology,2000,26(4):625-646.
[18]SAITOU N, NEI M.The neighbor-joining method:a new method for reconstructing phylogenetic trees.Mol Biol Evol,1987,4(4):406-425.
[19]ZHANG X Y, FANG Z W, ZHANG C F, et al.Effects of acarbose on the gut microbiota of prediabetic patients:a randomized,double-blind,controlled crossover trial.Diabetes Ther,2017,8(2):293-307.
[20]WU X K, MA C F, HAN L,et al.Molecular characterisation of the faecal microbiota in patients with type Ⅱ diabetes.Curr Microbiol,2010,61(1):69-78.
[21]ARUMUGAM M, RAES J, PELLETIER E, et al.Enterotypes of the human gut microbiome.Nature,2011,473(7346):174-180.
[22]DUNCAN S H, BELENGUER A, HOLTROP G, et al.Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces.Appl Environ Microbiol,2007,73(4):1073-1078.
[23]SATO J, KANAZAWA A, AZUMA K, et al.Probiotic reduces bacterial translocation in type 2 diabetes mellitus:a randomised controlled study.Sci Rep,2017,7(1):12115.
[24]HARTSTRA A V, BOUTER K E C, BCKHED F, et al.Insights into the role of the microbiome in obesity and type 2 diabetes.Diabetes Care,2015,38(1):159-165.
[25]KOOTTE R S, LEVIN E, SALOJRVI J, et al.Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition.Cell Metab,2017,26(4):611-619.e6.
[26]郭慧玲,邵玉宇,孟和畢力格,等.肠道菌群与疾病关系的研究进展.微生物学通报,2015,42(2):400-410.
(收稿日期:2021-10-19 修回日期:2021-12-29)
(编辑:潘明志)
猜你喜欢
山地农业生物学报(2022年3期)2022-05-13
土壤学报(2022年1期)2022-03-08
浙江农业学报(2022年1期)2022-02-18
学校教育研究(2020年7期)2020-04-09
大自然探索(2020年1期)2020-02-16
中学生理科应试(2017年6期)2017-09-27
上海预防医学(2017年8期)2017-09-06
百科知识(2016年16期)2016-10-29
现代家庭(2000年5期)2000-06-14
祝您健康(1988年5期)1988-12-30
