
Solvation Structure and Dynamics of Mg(TFSI)2Aqueous Electrolyte
2022-04-15 11:49:32ZhouYuTaylorJuranXinyiLiuKeeSungHanHuiWangKarlMuellerLinMaKangXuTaoLiLarryCurtissandLeiCheng
Energy & Environmental Materials 2022年1期
Zhou Yu ,Taylor R.Juran,Xinyi Liu,Kee Sung Han,Hui Wang,Karl T.Mueller,Lin Ma ,Kang Xu ,Tao Li*,Larry A.Curtiss*,and Lei Cheng*
Using ab initio molecular dynamics(AIMD)simulations,classical molecular dynamics(CMD)simulations,small-angle X-ray scattering(SAXS),and pulsed- field gradient nuclear magnetic resonance(PFG-NMR),the solvation structure and ion dynamics of magnesium bis(trifluoromethanesulfonyl)imide(Mg(TFSI)2)aqueous electrolyte at 1,2,and 3 m concentrations are investigated.From AIMD and CMD simulations,the first solvation shell of an Mg2+ion is found to be composed of six water molecules in an octahedral configuration and the solvation shell is rather rigid.The TFSI-ions prefer to stay in the second solvation shell and beyond.Meanwhile,the comparable diffusion coefficients of positive and negative ions in Mg(TFSI)2aqueous electrolytes have been observed,which is mainly due to the formation of the stable[Mg(H2O)6]2+complex,and,as a result,the increased effective Mg ion size.Finally,the calculated correlated transference numbers are lower than the uncorrelated ones even at the low concentration of 2 and 3 m,suggesting the enhanced correlations between ions in the multivalent electrolytes.This work provides a molecular-level understanding of how the solvation structure and multivalency of the ion affect the dynamics and transport properties of the multivalent electrolyte,providing insight for rational designs of electrolytes for improved ion transport properties.
Keywords
Mg(TFSI)2aqueous electrolyte,molecular dynamics simulation,pulsed- field gradient nuclear magnetic resonance,small-angle X-ray scattering,ion dynamics
1.Introduction
Multivalent-ion batteries play an important role in the quest for energy-dense alternatives to Liion batteries(LIBs).Since the 1970s,lithiumbased battery chemistries have been sought after as the ultimate solution to energy storage needs,with the eventual fruition of LIBs as the dominant component of current energy storage technologies.However,as the demand for an energy storage device of high energy,high safety,low-cost,and risk-free supply chain becomes ever imminent,battery chemistries based on earth-abundant elements rather than lithium emerge as candidates,targeting applications in personal electronics,vehicles,and grid storage.In the early 2000s,bivalent Mg was reported as such a promising candidate to substitute for Li.[1]The Mg battery concept brings benefits such as its divalent nature,which provides two electrons per redox center with a much higher theoretical volumetric capacity(3832 mAh cm-3)compared with monovalent ions such as Li+(2062 mAh cm-3)and Na+(1129 mAh cm-3).[2]However,the development of a functional Mg battery has encountered challenges in every component.Specifically,to Mg electrolytes,slow ion transport,limited electrochemical stability,and concomitant safety are key performance metrics that need to be improved.[3,4]
Aqueous electrolytes have drawn increasing attention over the past few years,due to the contribution of water in electrolytes to activate the electrode and help the Mg2+insertion process[5]and the significantly improved electrochemical stability brought by the“water-in-salt”concept.[6]Chen et al.[7]proposed an aqueous Mg-ion battery composed of a Prussian blue-type nickel hexacyanoferrate cathode,polyimide anode,and 1M(molarity,molsoluteper Lsolution)MgSO4aqueous electrolyte.In addition to achieving the maximum cell voltage of 1.5 V and the comparable operating voltage to the unstable non-aqueous electrolyte,the battery cell maintains 60% capacity after 5000 cycles,with almost 100% Coulombic efficiency.Furthermore,a~40 Wh kg-1energy density was achieved at a 1 A g-1current.[7]Wang et al.[8]proposed a highly concentrated aqueous Mg-ion battery composed of Li3V2(PO4)3cathode,polypyromellitic dianhydride anode,and 4 m (molality,molsoluteper kgsolvent)magnesium bis(trifluoromethanesulfonyl)imide(Mg(TFSI)2)aqueous electrolyte.The stable electrochemical window is increased to 2.0 V.Additionally,92% initial capacity can be retained after 6000 cycles.Moreover,an unprecedented high power density of 6400 W kg-1was achieved,which is~30 times higher than the non-aqueous Mg-ion batteries in the literature.[8]
The cation solvation structure and dynamics in liquid electrolytes dictate critically important properties essential to battery performance.For example,the ion-pairing can impact the ion transport and charge transfer,[9]while the solvation structure often predicts the interfacial chemistry and,consequently,the electrochemical stability.[6]The use of aqueous Mg electrolytes is still in its infancy and only limited efforts were reported.Buchner et al.[10]found the effective hydration number,including the first and second solvation shells around the Mg2+ion,to decrease as the Mg(SO4)2concentration is increased.Wahab et al.[11]proposed that ion pairs do not form in the 1MMg(NO3)2aqueous solution.Recently,Bucur et al.[4]proved that the hydration of Mg ion might benefit the intercalation process in oxides.
While these elegant works explored the fundamental physicochemical properties of Mg aqueous electrolytes,many key questions to the design of these electrolytes,such as their structural and dynamical properties,remain unanswered.In this work,we aimed to study the relationship between the solvation and dynamics in divalent(i.e.,Mg2+ion)aqueous solutions and compare these properties with monovalent(i.e.,Li+ion)solution.We utilize ab initio and classical molecular dynamics(MD)simulations,small-angle X-ray scattering technique(SAXS),and pulsed- field gradient nuclear magnetic resonance measurements(PFG-NMR)to investigate the multivalent and concentration effects on the solvation structure and dynamics of the Mg(TFSI)2aqueous electrolyte.We found that a rigid octahedral hydration shell around the Mg2+ion leads to a significant cage effect at the ps timescale.Meanwhile,the long lifetime hydration shell around Mg2+ion and similar ion size of[Mg(H2O)6]2+and TFSI-ion could be the decisive factors in the comparable diffusion coefficients of cations and anions in Mg(TFSI)2aqueous electrolyte for various concentrations.Furthermore,we found the transport properties of electrolytes at high concentrations deviate from the Einstein relation.
2.Results and Discussion
2.1.Solvation Structure
While a recent study has demonstrated that nearly all ions maintain the solvent-separated single-ion state in the 1 m LiTFSI aqueous electrolyte,[12]clear evidence of the solvation state of the ion in the divalent Mg(TFSI)2aqueous electrolyte remains absent in the literature.The initial configuration plays an important role in the AIMD simulations due to the limited simulation timescale used(i.e.,~100 ps).A carefully set up initial configuration helps the system reach a meaningful configuration faster.We performed AIMD simulations with two distinct initial salt configurations:solvent-separated single-ion and contacting-ion pair,respectively,to elucidate the ion behavior.The solvent-separated single Mg2+ion system maintains its configuration for the entire simulation(~50 ps).Conversely,in the simulation starting with the contacting ion pair configuration,we observe the Mg2+ion dissociating from the TFSI-ion(Figure 1a)within~12 ps of simulation time.These results suggest that ions prefer the solvent-separated single-ion state in the 1 m Mg(TFSI)2aqueous electrolyte.Therefore,the following analysis of the structure and dynamics was conducted for the solvent-separated single-ion structure for both the Li+and Mg2+ion systems.

Figure 1.AIMD simulation results of 1 m LiTFSI and 1 m Mg(TFSI)2aqueous electrolytes.a)Time evolution of the distance between Mg2+ion and the coordinated oxygen from TFSI-ion.b)Radial distribution function g(r)and coordination number N(r)of water molecules around Li+and Mg2+ions.c)Histogram of the angle formed between oxygen in water,cation,and oxygen in the water.The probability ratio of the two marked peaks for the Mg case is 4:1.d)Potential of mean force of water molecules as a function of their distance from Li+and Mg2+ions.The black arrow between the dashed lines indicates the energy barrier for a water molecule to escape from the first hydration shell around Li+ion.
To characterize the multivalent effects on the solvation structure around the cation,we compute the radial distribution function(RDF)and the coordination number of water molecules surrounding Li+and Mg2+ions(Figure 1b).In the Li-1S system,the first hydration shell is composed of 4 water molecules at a distance of~0.20 nm from the cation.The water molecules at ~0.40 nm from the Li+ion compose the second hydration shell,which corresponds to a much broader RDF peak than the first one in Figure 1b.In the Mg-1S system,the first hydration shell is composed of 6 water molecules at a slightly further distance of 0.21 nm.A distinct second hydration shell represented by another sharp peak is located at~0.43 nm radially from the Mg2+ion.The analysis on the histogram of the angles formed by oxygen in water,cation,and oxygen in the water of the first hydration shell(Figure 1c)shows that the mean angle is 109.0± 10.8°in the Li-1S system,which indicates that the four water molecules in the first hydration shell form a tetrahedral coordination environment.In the Mg-1S system,there are two peaks at angles of 90.0 ± 6.3°and 170.7± 5.0°,respectively,with the probability ratio of 4:1.This angle distribution suggests an octahedral coordination configuration around the cation in the first hydration shell.These observations are consistent with previous studies.[13]Furthermore,the Mg-1S system possesses a smaller full width at half maximum(FWHM)and a reduced standard deviation of angle distribution compared with the Li-1S system.This suggests that the oscillation of water molecules in the first hydration shell surrounding Mg2+ion is rather confined.In other words,the hydration shell around the Mg2+ion is more rigid compared with that surrounding the Li+ion.
It is also essential to understand the thermodynamics of the exchange of water molecules around the cations,which can affect the intercalation process and dynamic properties.[14]We also computed the potential of mean force(PMF),which reflects the potential energy change in water molecules as a function of its distance from the cation.It is computed using PMF( r)=-kBTln[g( r)],where kBis Boltzmann’s constant,T is the absolute temperature,and g(r)is the RDF from the targeted cation to the water molecules.The water molecule in the first hydration shell around the Li+ion needs to overcome an energy barrier of~17 kJ mol-1(the black arrow in Figure 1d)to migrate to a neighboring hydration shell.No exchanges of water molecules occur between the two hydration shells at ~0.20 and ~0.43 nm surrounding the Mg2+ion over the course of the simulation period,which implies that a water molecule within the first hydration shell surrounding an Mg2+ion must overcome a larger energy barrier to migrate to a neighboring shell than that required for a hydrated Li+ion system.
付江录是十三师火箭农场的个体工商户。他先后成立了哈密江盛有限责任公司、哈密鼎舜有限责任公司,在此期间,他还从事过个体运输和机采棉等工作。无论在何种岗位,他都是干一行爱一行,一心扑在工作上,兢兢业业,勤勤恳恳,一丝不苟,各项工作想在前、干在前,充分起到了模范带头作用。他用自己200多万元的资金帮扶了近40人脱贫致富,其中有8名火箭农场的少数民族兄弟在他的倾情支持下,从一无所有踏上了小康之路。
Furthermore,to characterize the charge-delocalization effects between the cation and the hydration shell,we performed Bader’s charge analysis[15-18]on one hundred frames that were extracted from a 10 ps trajectory of each simulation with a time interval of 100 fs.We found the Li and Mg ions had 0.90 and 1.75 positive charges,respectively.Meanwhile,the first hydration shell of the Li and Mg ions had 0.11 and 0.19 negative charges,respectively.Considering the distance between cation and O in water shown in Figure 1b,we can estimate the electrostatic interaction potential between the ion and hydration shell(Ecation-water)following Ecation-water=ke(qcationqwater/r),where keis Coulomb’s constant,qcationand qwaterare the atomic charges of the cation and hydration shell,respectively,and r is the distance between the cation and hydration shell.The ratio of EMg-waterto ELi-wateris~3.The strong electrostatic interaction between Mg2+ion and first hydration shell contributes to the high solvation energy of Mg2+ion is shown in Figure S2.Meanwhile,this observation is in line with our observation on the PMF in Figure 1d.That is,water molecules in the first hydration shell around an Mg2+ion need to overcome a much larger energy penalty in order to escape from this shell than that required for a hydrated Li+ion system.
The dynamics of ions in high concentrations are sluggish and AIMD simulations are computationally expensive.Therefore,we use the CMD simulations to study the concentration effects on the structure and dynamics of Mg(TFSI)2aqueous electrolytes.The force field used in our CMD simulations is firstly validated against the structural property results from our AIMD simulations and experiments.From Figure 2a,we see that the RDF and the coordination number of oxygen atoms from water molecules around the Mg2+ion in Mg-1 system,calculated with CMD simulation,are consistent with those calculated with AIMD simulation in Mg-1S system.There is a slight difference of 0.01 nm on the location of the first peak in the RDF from Mg2+ion to the oxygen in water obtained from the two methods.Furthermore,the structure factor shifts calculated with CMD simulation have similar trends to the experimental SAXS data across the three different concentrations,as shown in Figure 2b1,b2.These results show that the OPLS-aa force fields in CMD capture the main structural features of Mg(TFSI)2aqueous electrolytes.Further decoupling of the structure factor by the interactions between different components of the electrolyte can be found in Figure S3.

Figure 2.a)Radial distribution function g(r)and coordination number N(r)of oxygens in water around Mg2+ion in 1 m Mg(TFSI)2aqueous electrolyte calculated using AIMD and CMD.Structure factors of the 1,2,and 3 m aqueous electrolytes b1)are measured using SAXS and b2)calculated using CMD simulation.
Using CMD,we analyzed the solvation environments of Mg2+ions,including water molecules and TFSI-ion,at various concentrations(Figure 3).We see two distinct hydration shells within a 0.5 nm radius of the Mg2+ion.The first hydration shell is composed of 6 water molecules and is maintained at all three concentrations considered.The radial density and coordination number of water molecules in the second hydration shell decrease as the salt concentration is increased.This is due to TFSI-ion appearing at~0.5 nm and replacing water molecules in the second hydration shell at increased concentrations(Figure 3b).The radial distribution and coordination number of oxygen atoms in TFSI-ion increase as the concentration of salt increases.We find two prominent peaks at 0.44 and 0.68 nm,respectively,in the radial density distribution of oxygen atom in TFSI-ion.We note that the two peaks correspond to the two oxygen atoms binding to the same sulfur in TFSI-ion(Figure 3b).
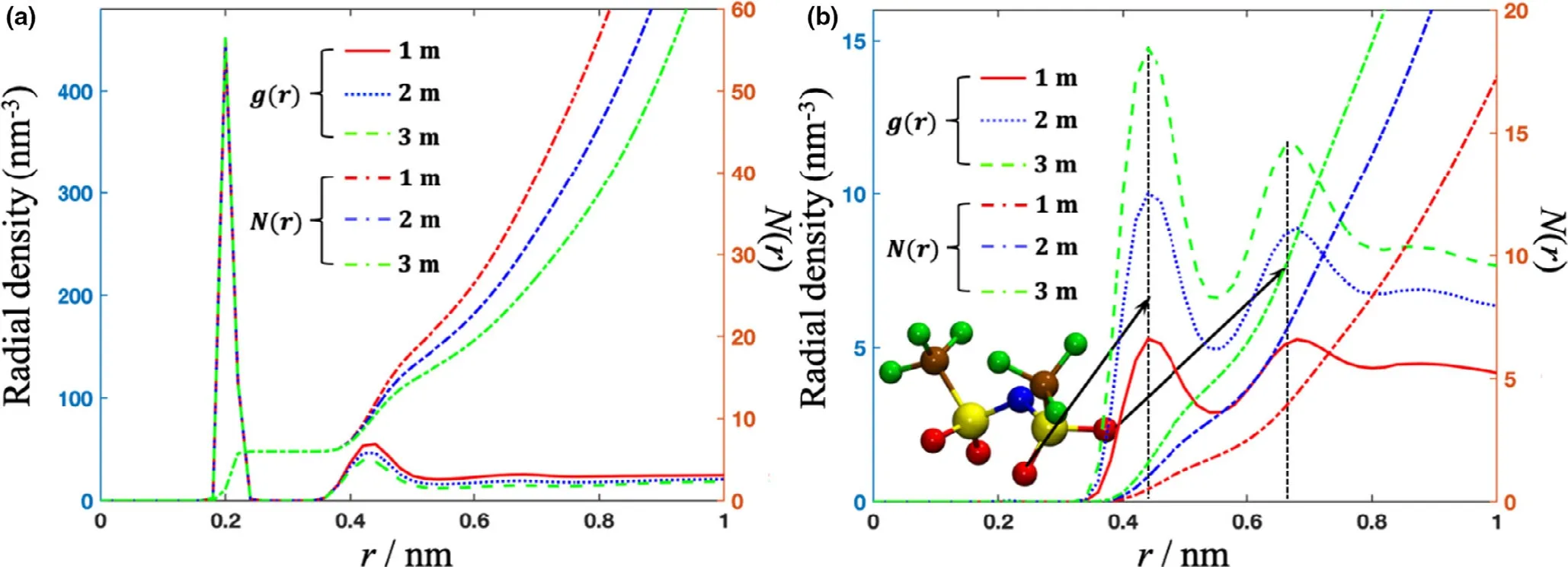
Figure 3.Radial density distribution and coordination number of a)oxygens in water and b)oxygens in TFSI-ion around Mg2+ion in 1,2,and 3 m aqueous electrolyte calculated using CMD simulation.The two vertical black dashed lines in Figure 3 guide the locations of the two peaks.The inset TFSI-ion shows the two oxygen atoms binding with the same sulfur attributing to the two peaks.Blue,yellow,brown,green,red,and white balls denote the N,S,C,F,O,and H atoms.
2.2.Dynamics
There are two theoretically equivalent methods to quantify the self-diffusivity of MD simulation systems,which are known as the Green-Kubo method and the Einstein method.In practice,the Einstein method is often preferred due to the precision and reproducibility of the post-simulation data analysis.[19]The general procedure of the Einstein method is to calculate the slope of the diffusive regimes in the mean-square displacements(MSDs)versus time plot.Unfortunately,within the AIMD simulation timescale of less than~100 ps,the dynamics of many systems do not reach the diffusive regimes that are meaningful for the self-diffusivity calculations.[20]We use a statistical parameter introduced by Burov et al.[21]to distinguish the dynamics of Li+and Mg2+ion at 1 m concentration within the relatively short AIMD trajectories we have.Specifically,three successive coordinates of the targeting ion,with a time interval Δ, are used to define two displacement vectors:V1(t)=X(t+Δ)-X(t);V2(t)=X(t+2Δ)-X(t+Δ),where X(t)is the coordinate of a targeted ion at time t.A small angle between V1(t)and V2(t)would indicate that the ion moves along almost the same direction as the previous time step,and the movement is barely restricted by the solvation shell.We can consider such trajectories as forward ballistic motion.A large angle between V1(t)and V2(t)would indicate the ion movement changed its direction,because the forward motion is blocked by other atoms occupying the position.This means a cage effect of the solvation shell is prominent.
We compute the ensemble average of the angle between these two displacement vectors as a function of the time interval for the two systems,and the results are shown in Figure 4a-d.Specifically,the angles between displacement vectors at different time t from the simulation trajectory were calculated and then used to obtain the average at a given time interval Δ.A similar angle distribution for Li+and Mg2+ion has been observed within 1 ps(Figure 4a,c).Initially,a high frequency on the distribution occurs around the small angles,which suggests that the cation maintains forward ballistic motion.The histograms of the angle probability distribution for the time interval of 10 fs are shown in Figure 4e.We can see that the ion does not undergo the Brownian motion(random walk of particles),which would correspond to an equal distribution at all angles,as indicated by the horizontal dotted line.As the angle distribution increases from 0 to ~180 degrees in ~0.4 ps,as guided by the white dashed line in Figure 4a,c,we depict the transition from forward ballistic to motion that is affected by the solvation cage,which limits the forward movement of the ion.The angle distribution of Li+and Mg2+ions stabilizes around 110 and 140 degrees in 8 ps,respectively(Figure 4f).On average,the angle distribution in the Mg-1S system is higher than the Li-1S system,suggesting that the rigid octahedral hydration shell surrounding Mg2+ion leads to a stronger cage effect,which hinders the ionic motion.

Figure 4.Histograms of the displacement vector angle probability distribution as a function of the time interval Δ for the a)0-1 ps and b)1-10 ps in the Li-1S system,as well as c)0-1 ps and d)1-10 ps in the Mg-1S system from AIMD simulations.Histograms of angle probability distribution with a time interval of e)10 fs and f)8 ps.BM stands for the Brownian motion.White dashed lines in a,c)indicate the transition trend from forward ballistic motion to the cage effect dominating motion.White dotted lines in a-d)represent a time interval of 10 fs and 8 ps.The horizontal dotted line in e,f)represents the distribution of Brownian motion for comparison.
CMD was used to explore the concentration effects on the dynamics of Mg(TFSI)2aqueous electrolyte.The lifetime of the water and TFSI-ion in the first two hydration shells was characterized by calculating the residence correlation function for water molecules within the first and second hydration shells,the oxygen atoms in TFSI-ion,and the TFSI-ion in the second hydration shell.The residence correlation function is defined as <c( 0)c( t)>,where c( t)is an indicator of the targeted particle’s category(hydration shell).The c( t)is defined as 1.0 if the targeted particle in the corresponding shell at time t=0 resides continuously in this shell by time t,and 0 if the atom or molecule is dissociated from the ion according to the cutoff distances defined by the valleys in the RDF plot.For the definition of the residence time of the TFSI-ion,as long as one of the four oxygen atoms from the ion remains in the solvation shell,we consider the ion resides.In Figure 5,we plot out the time evolution of the statistical average residence correlation function for all cations in the simulation box with a time interval of 1 ps.A rapid decay in correlation function indicates that the targeted particle has a stronger tendency to escape from the corresponding hydration shell.From Figure 5a-c,we see the residence correlation functions for water and TFSI-ion in the first two hydration shells are similar for the three concentrations.Water molecules in the first hydration shell do not leave the Mg2+ion throughout the timescale used for our simulations(~30 ns).This observation is consistent with a previous NMR study,which reports an exchange timescale for water on the order of microseconds for Mg2+in the first hydration shell.[22]Furthermore,we see that when compared with water molecules,the coordinated oxygen atoms in the TFSI-ion from the second hydration shell escapes from Mg2+ion more readily.However,the dissociation of one oxygen is usually replaced by another oxygen atom from the same TFSI-ion.As a result,the TFSI-ion in the second hydration shell“resides”for a longer time than the water molecule in the first and this is evidenced by the slower decay of TFSI-ion on the residence correlation function.If we define the relaxation time as the time it takes for <c( 0)c( t)> to decrease to 5% ,we see that the relaxation time increases as the concentration increases(Figure 5d).Interestingly,the concentration affects the relaxation time of TFSI-ion more pronouncedly than that of the water molecules,as evidenced by the steeper slope for the relaxation time of TFSI-ion as a function of concentration.

Figure 5.Residence correlation function for water in the first(r<0.30 nm in Figure 3a)and second(0.30 nm<r<0.50 nm in Figure 3a)hydration shells,the TFSI-ion and the oxygen atom in TFSI-ion in the second(0.30 nm<r<0.50 nm in Figure 3b)hydration shell in a)1,b)2,and c)3 m concentration Mg(TFSI)2aqueous solutions.d)Relaxation time defined as the time when <c( 0)c( t)> =0.05 as a function of concentration.
Then,we calculated the diffusion coefficients of Mg2+,TFSI-ions,and water molecules as a function of salt concentration from the CMD simulations using the Einstein method.From Figure 6a,we can see the diffusion coefficients of ions and water decrease with the increase in salt concentration.The diffusion coefficients of Mg2+and TFSI-are comparable within the error bars,and water diffusion is about 3-4 times faster than for both Mg2+and TFSI-for various concentrations.All these critical features have also been captured in the PFG-NMR experiments(Figure 6b).The error in the diffusion coefficient of Mg2+ions is slightly larger than that of TFSI-anions and H2O molecules due to the lower signal intensity,that is,the signal-to-noise ratio in the25Mg PFG-NMR(inset of Figure S1).Here,we note that the diffusion coefficients calculated in MD simulation are 3-4 times larger than those measured in experiments.This is mainly because the TIP3P force field and the small scaling factor on the atomic charges in the force field of ions(i.e.,0.8)can significantly overestimate the mobility of water molecules and ions.The diffusion coefficients were also calculated in the MD simulations with a scaling factor of 0.9 and 1.0 on the atomic charges in the force fields of ions(see Figure S4).The key features,such as the high diffusion coefficient of water molecules and comparable diffusion coefficient of cation and anion,remain.
The similar diffusion coefficients of the anion and cation in the aqueous Mg(TFSI)2electrolytes differ from that in the LiTFSI aqueous electrolyte,in which the diffusion coefficient of Li+is 50-130% larger than that of the TFSI-ion at 1-21 m concentrations using CMD simulations and PFG-NMR measurements.[12,23]This feature can be explained through the electrostatic interaction and ion size.The strong electrostatic field exerted by the divalent cation leads to a strong correlation between cation and anion.Therefore,the diffusion coefficient differences should be less than those in the monovalent electrolyte with similar concentration.Considering that the rigid octahedral hydration shell has a relatively long lifetime,the Mg2+ions in the electrolyte exist as[Mg(H2O)6]2+complex cations,which increases the “effective”ion size.The size of the first hydration shell around Mg2+ion(i.e.,0.4-0.5 nm)is similar to the distance between carbon atoms in one TFSI-ion(inset of Figure 3b),which also contributes to the comparable dynamics of ions.

Figure 6.Diffusion coefficient of Mg2+,TFSI-,water,and the ratio of Mg2+and TFSI-as a function of concentration calculated in a)simulation and b)measured through PFG-NMR.Diffusion coefficients of TFSI-and water were calculated based on the center of mass of TFSI-ion and the oxygen atom in a water molecule in a CMD simulation.c)Uncorrelated and correlated transference numbers(t+uncand t+c)calculated in the CMD simulations and the transference number measured through PFG-NMR.d)The diffusion coefficient and viscosity relationship.Three points represent the experimental diffusivity and viscosity,and the black curve was calculated based on the Stokes-Einstein relations by taking the experimental diffusivity and viscosity of 1 m electrolyte as the reference.


Furthermore,we measured the viscosities of the electrolytes and explored diffusion coefficient-viscosity relations.First,we estimated the diffusion coefficient of the electrolytes by the approximation D=xcDc+xaDa+xwDw,where xc,xa,xw,Dc,Da,and Dwrepresent the mole fractions and experimental diffusion coefficients of cation,anion,and solvent,respectively.[28]If the electrolyte is in the dilute regime and the correlation between ions can be ignored,the ions can be viewed as independent in their respective motions,and the Stokes-Einstein relation holds as ηiDi=ηjDj,where ηi,ηj,Di,and Djrepresent the viscosity and diffusion coefficient of the two systems i and j,respectively.By taking the experimental diffusion coefficient and viscosity of 1 m electrolyte as the reference,we can calculate the relationships between the viscosity and diffusion coefficient shown as the black curve in Figure 6d.However,the experimental viscosity deviates from the Stokes-Einstein relation as the concentration increases.For example,the Stokes-Einstein relation underestimates the viscosity by~2.3% and~7.3% at 2 and 3 m,respectively,due to ignoring the ionic correlations.These observations on the correlated transference number and viscosity once again remind us to exercise caution when applying these transport laws,which were originally derived for dilute/ideal electrolytes,to the highly concentrated regimes.
3.Conclusion
Using AIMD and CMD simulations,SAXS,and PFG-NMR,we investigated the solvation structure and dynamics of the cation in the divalent Mg(TFSI)2aqueous electrolyte and in the LiTFSI aqueous electrolyte for comparison.With AIMD,we found the octahedral hydration shell composed of 6 water molecules around Mg2+ion is more rigid than the tetrahedral shell composed of 4 water molecules around Li+ion.The strong electrostatic interaction between Mg2+ion and hydration shell is an important component of the high solvation energy and results in slow water exchange between hydration shells.Using experimentally verified CMD,we found TFSI-ions can only exist in the second solvation shell or beyond.The dynamics of 1,2,and 3 m Mg(TFSI)2aqueous electrolytes were further studied using CMD simulations.We found that the two oxygen atoms from the same TFSI-ion can interexchange;the TFSI-ion itself does not escape the second solvation shell readily.Unlike the LiTFSI electrolyte in which the cation dynamic is faster than the anion,the diffusion coefficients of the Mg cation and the anion are comparable.This is due to the formation of the stable[Mg(H2O)6]2+complex,resulting in the increased effective size that slows down the cation.As expected,the diffusion coefficient of water is significantly higher than the diffusion coefficients of both ions.Remarkably,the calculated correlated transference numberis much smaller than the uncorrelatedeven at the low concentrations of 2 and 3 m.This highlights the highly nonideal and correlated nature of the multivalent salts.
This work has contributed to the understanding of the solvation structure and the resulting dynamic behaviors in the multivalent electrolytes.We showed that the strong electrostatic interaction between a multivalent ion and its negatively charged solvation shell and the counterion leads to the enlarged effective size and strong correlation of the Mg2+ion,ultimately slowing down the ion diffusion and decreasing correlated transference number.A further design goal for fast ion-conducting multivalent electrolytes should focus on achieving a moderate solvent-ion interaction and,consequently,a more flexible solvation shell through careful selections of solvents or diluents.
4.Computational and Experimental Methods
Ab Initio/Classical MD Simulations:Ab initio molecular dynamics(AIMD)simulations were performed on 1 m LiTFSI and 1 m Mg(TFSI)2aqueous electrolyte models,composed of 1 salt molecule and 55 water molecules.Classical MD(CMD)simulations were performed on 1,2,and 3 m Mg(TFSI)2aqueous electrolyte solutions with 90,180,and 270 salt molecules in the simulation models,respectively.Initial configurations were first set up using Packmol code.[29]The simulation box is periodic in all three directions.Additional details of the AIMD and CMD system setups are summarized in Table 1.

Table 1.Setup of the AIMD and CMD systems studied in this work.
The AIMD simulations were carried out using the Born-Oppenheimer approximation with the VASP code.[30-32]The projector augmented wave(PAW) method[33]was used to compute the interatomic forces with the Perdew--Burke-Ernzerhof(PBE)generalized-gradient approximation[34]for the exchangecorrelation energy.The plane-wave energy cutoff was set as 500 eV,and the Brillouin zone was sampled at the Γ-point.All AIMD simulations were performed with the NVT ensemble.The Nosé-Hoover thermostat[35]was used to stabilize the temperature of the simulations at 298 K.A time step of 0.5 fs was utilized for all simulations.Finally,simulations were run for a total time of 60 ps.The initial 20 ps trajectory was used to equilibrate the system.The standard deviation of the system energy during the last 5 ps of the equilibration run is less than 1 meV atom-1ps-1,which is small enough that the system can be considered to reach equilibration.[36]The 40 ps trajectory that follows was then considered as the “production”run,and the results were used for analysis.
The CMD simulations were performed using GROMACS 5.1.4 with a 2 fs time step.[37]The bonded and non-bonded parameters of the force fields for Li+,Mg2+,and TFSI-ion were obtained from previous studies,[38,39]which were developed and optimized within the framework of an OPLS-aa force field.[40]The atomic charges in the force field of ions were scaled by 0.8 to compensate for the charge transfer,polarization,and binding effects.[41]The TIP3P model was employed for the water molecules.[42]Simulations were initially equilibrated under an NPT ensemble for 20 ns.Then,the production simulations were executed with an NVT ensemble for 250 ns.The temperature of the system was maintained at 298 K using the velocity-rescaling thermostat.[43]Non-electrostatic interactions were computed by direct summation with a cutoff length of 1.2 nm.Electrostatic interactions were computed using the particle mesh Ewald(PME)method.[44]The real space cutoff and Fourier spacing were 1.2 and 0.12 nm,respectively.The box size and periodic boundary condition of the CMD simulations have been evaluated by comparing the results of simulations with a variety of cell sizes.
SAXS Measurements:Mg(TFSI)2was purchased from Sigma-Aldrich.Aqueous electrolytes were prepared by molality(molsoluteper kgsolvent).SAXS experiments were performed at the Advanced Photon Source(APS)12ID-B and C station of Argonne National Laboratory.The 2D SAXS data were collected on a PILATUS 2 M area detector(Dectris Ltd.)with a 2 meter distance from the sample to the detector and the incident energy of 13.3 keV.The two-dimensional scattering images were radially averaged over all orientations to produce plots of scattered intensity I(q)versus scattering vector q,where q=4π sin θ/λ.The scattering vector was calibrated using silver behenate.The samples were loaded into 1.5 mm diameter quartz capillary tubes for the SAXS measurements.
PFG-NMR Measurements:PFG-NMR measurements were performed on a 600 MHz NMR spectrometer(Agilent,USA)with a 5 mm liquid NMR probe(Doty Scientific,USA),which can generate a z-gradient up to~31 T m-1.Diffusion coefficients of Mg2+cations,TFSI-anions,and H2O molecules were determined from25Mg,19F and,1H PFG-NMR,respectively,at 25°C.All diffusion coefficients were measured using the bipolar gradient stimulated echo sequence(Dbppste,vendor-supplied sequence,VNMRJ,Agilent).The PFG-echo profiles were recorded as a function of gradient strength with 16 equal steps and fitted with the Stejskal-Tanner equation:[45]S(g)=S(0)exp[-D(γgδ)2(Δ - δ/3)],where S(g)and S(0)are the echo heights at the gradient strengths of g and 0,D is the diffusion coefficient,γ is the gyromagnetic ratios of25Mg,19F or1H,Δ is the diffusion delay,which is the time interval between the two pairs of bipolar pulse gradients,and δ is the gradient length.The time interval Δ was 15 ms for25Mg PFGNMR and 40 ms for both1H and19F PFG-NMR.The gradient length δ was 2 ms for all measurements.The maximum gradient strength required for25Mg PFGNMR was~19 T m-1(see Figure S1),which is stronger approximately 16 times than that of1H/19F PFG-NMR(~1.2 T m-1)due to the fact that γ(1H)/γ(25Mg)≈16.4.
Viscosity Measurements:Viscosity was measured using an Ostwald viscometer(Sibata Scientific Technology,Japan)with a capillary diameter of 0.75 mm.The temperature of the electrolyte in the viscometer was controlled by a circulating water bath(Sibata Scientific Technology,Japan)at 25°C.
Acknowledgements
This research was supported by the Joint Center for Energy Storage Research(JCESR),a U.S.Department of Energy,Energy Innovation Hub.The submitted manuscript has been created by UChicago Argonne,LLC,Operator of Argonne National Laboratory(“Argonne”).Argonne,a U.S.Department of Energy Office of Science laboratory,is operated under Contract no.DE-AC02-06CH11357.This research used resources of the Advanced Photon Source,a U.S.Department of Energy(DOE)Office of Science User Facility,operated for the DOE Office of Science by Argonne National Laboratory under Contract No.DE-AC02-06CH11357.The PFG-NMR measurements were performed at the Environmental Molecular Sciences Laboratory(EMSL),a national scientific user facility,sponsored by the DOE’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory(PNNL).We acknowledge Dr.Yuyue Zhao for the density measurement of Mg(TFSI)2aqueous electrolytes.We gratefully acknowledge the computing resources provided on Bebop,a high-performance computing cluster,operated by the Laboratory Computing Resource Center at Argonne National Laboratory.
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
猜你喜欢
办公室业务(2018年3期)2018-04-12 00:33:20
棉花科学(2017年1期)2017-03-10 20:38:43
企业文化·下旬刊(2016年5期)2016-06-04 17:31:54
中国棉花加工(2015年4期)2015-12-19 06:40:56
湖北农业科学(2015年16期)2015-10-28 21:15:39
传奇故事(破茧成蝶)(2015年8期)2015-02-28 09:29:34
中国火炬(2014年8期)2014-07-24 14:30:18
作文周刊·小学一年级版(2014年34期)2014-04-29 00:44:03
中国集体经济·下(2013年6期)2013-04-29 00:44:03
中国火炬(2010年2期)2010-07-24 14:36:02
 Energy & Environmental Materials2022年1期
Energy & Environmental Materials2022年1期
- Energy & Environmental Materials的其它文章
- Introduction of Frontier Outlook
- Sn Alloy and Graphite Addition to Enhance Initial Coulombic Efficiency and Cycling Stability of SiO Anodes for Li-Ion Batteries
- Biomass Template Derived Boron/Oxygen Co-Doped Carbon Particles as Advanced Anodes for Potassium-Ion Batteries
- A Stretchable Ionic Conductive Elastomer for High-Areal-Capacity Lithium-Metal Batteries
- Gas Generation Mechanism in Li-Metal Batteries
- Understanding the Coffee ring Effect on Self-discharge Behavior of Printed micro-Supercapacitors