
深度学习模型在中药毒性预警中的应用和前景
2022-04-13 02:50颜彩琴范睿琦宁雨坪
中国药理学与毒理学杂志 2022年3期
颜彩琴,范睿琦,宁雨坪,郭 宪,王 凯
(1.天津中医药大学中药学院,天津 301617;2.南开大学人工智能学院,天津 300350)
中药潜在毒性成分的预警在中药新药研发、安全使用及风险防控决策等方面具有重要意义。目前用于药物毒性成分预警的方法和手段主要包括实验方法和计算机预测方法。实验方法主要是在实验室中进行动物实验及体外细胞实验,耗损率高,周期长,经济效益低[1]。计算机预测方法是利用机器学习模型直接对药物毒性进行预测,无需进行大量实验,经济效益高[2]。近年来,随着训练数据的增多,机器学习技术得到快速发展。
早期用于毒性预警的机器学习方法是将药物分子进行编码,然后输入到传统机器学习算法架构的模型中,常见的有支持向量机(support vector machine,SVM)模型、随机森林(random forest,RF)模型、朴素贝叶斯模型、决策树模型和k-近邻模型。传统机器学习模型的优点是需要的数据量少,模型训练快,然而其表征能力有限,预测准确度较低[3]。随着深度学习(deep learning,DL)算法,也称为深度神经网络(deep neural network,DNN)的发展,DL模型逐渐替代传统机器学习模型用于药物毒性的预测[4]。与传统机器学习模型相比,DL模型通过构建多层人工神经网络(artificial neural network,ANN),从大量的数据中自动学习毒性化合物的结构特征,其准确性和可靠性得到极大提升。本文综述了DL模型基于“结构预警子-毒性”关系预测化合物潜在毒性的机制以及在药物分子毒性预测和毒理学研究中的具体应用,并提出DL模型在中药毒性预警中的挑战和展望,以期借助人工智能技术保障上市中药临床应用的安全性。
1 中药致毒机制及深度学习模型
1.1 “结构预警子-毒性”关系
“结构预警子-毒性”关系是机器学习模型预测化合物毒性的基本依据之一。通过识别数据集中的化合物结构信息,机器学习模型能够提取出化合物与毒性相关的结构特征,从而预测目标化合物的毒性[5]。
某些基团被认为是许多有毒中药单体毒性的结构标志,如黄独素B[6]、补骨脂素[7]和薄荷呋喃[8]中的呋喃环,千里光碱[9]中的吡咯里西啶,马钱子碱[10]、吴茱萸碱[11]和吴茱萸次碱[12]中的吲哚环及小檗红碱[13]中的喹啉环等,这些基团被称为“结构预警子”(structural alert)、毒理团或毒性片段[14]。研究表明,具有“结构预警子”的化合物引发毒性的关键一步是“结构预警子”的氧化反应[15-16]。在肝细胞色素P450酶的催化下,“结构预警子”被氧化形成具有高度亲电性的反应性代谢产物(reactive metabolite,RM),这一过程被称为化合物的“代谢活化”。RM可能与体内重要生物大分子,如蛋白质、酶和DNA的亲核基团发生共价结合,进而引发一系列的毒性反应。因此,在中药研发早期,快速并全面筛查化合物中的“结构预警子”并预测RM的形成是中药毒理学研究的重要组成部分,对于规避中药引发临床毒性事件至关重要。
筛查化合物“结构预警子”的实验室方法,是在肝微粒体孵化体系中加入亲核试剂,如谷胱甘肽、N-乙酰半胱氨酸和N-乙酰赖氨酸作为RM的捕获剂,并借助液质技术,对捕获剂共价结合RM所形成的稳定加合物进行定性,从而间接推断出RM的结构和原化合物发生代谢活化的反应中心,即“结构预警子”[17-18]。然而,实验室方法耗时长且需要昂贵的检测仪器,并且离不开人工操作,难以在短期内实现大量化合物的检测。为此,“结构预警子”的概念自1985年被提出后,不同领域的专家将在自然界和合成化合物中发现的“结构预警子”汇编形成一套“专家系统”,用于直观、快速地评估化合物的毒性[19]。然而,由于基团之间的相互影响,并非所有具有“结构预警子”的化合物都发生代谢活化并致毒。所以,只根据“结构预警子”预测化合物的毒性可能会出现假阳性结果,而且无法评估其毒性强弱[20]。
以DL模型为代表,近年发展起来的的机器学习模型能够弥补“专家系统”方法的不足。事实上,两者结合能获得更准确的预测结果。一方面,DL模型具有多层神经网络,能够综合分析整个分子结构而非局限于某个毒性基团,从而提取出超越“结构预警子”的高层特征。另一方面,在建立数据集时,以“结构预警子”作为输入特征,能更好地训练DL模型对隐性特征的提取和组合能力[21-22]。因此,在药物毒性预测中,与基于统计学的“专家系统”方法相比,基于“结构预警子-毒性”关系的DL模型可能获得更高的识别率。
1.2 深度学习模型的开发
在中药的毒性预测中,DL模型的基本开发流程如图1所示。首先是根据化合物的结构或生物学特征对筛选出的化合物进行分类,然后使用分子描述符将化合物的信息字符化,生成带毒性标签的训练集并输入到合适的DNN架构中。DL模型通过逐层特征变换自动对化合物的数据进行毒性表征,从而拥有预测化合物毒性的能力[23-24]。经过验证,若模型表现出较佳的性能,则可应用于中药毒性预警。

图1 预测中药毒性成分深度学习(DL)模型的开发流程[24].
1.2.1 毒性化合物数据集的建立
足够数量和高质量的数据是DL模型性能的保证[24],如数据量少且多样性低下,则可能导致DL模型泛化不足,对数据产生偏向性。表1列举了常用的毒性化合物数据库。为保证数据集的质量,在输入模型之前,还需对采集的数据进行筛选,过滤掉错误的以及重叠的数据,并对化合物结构进行标准化。
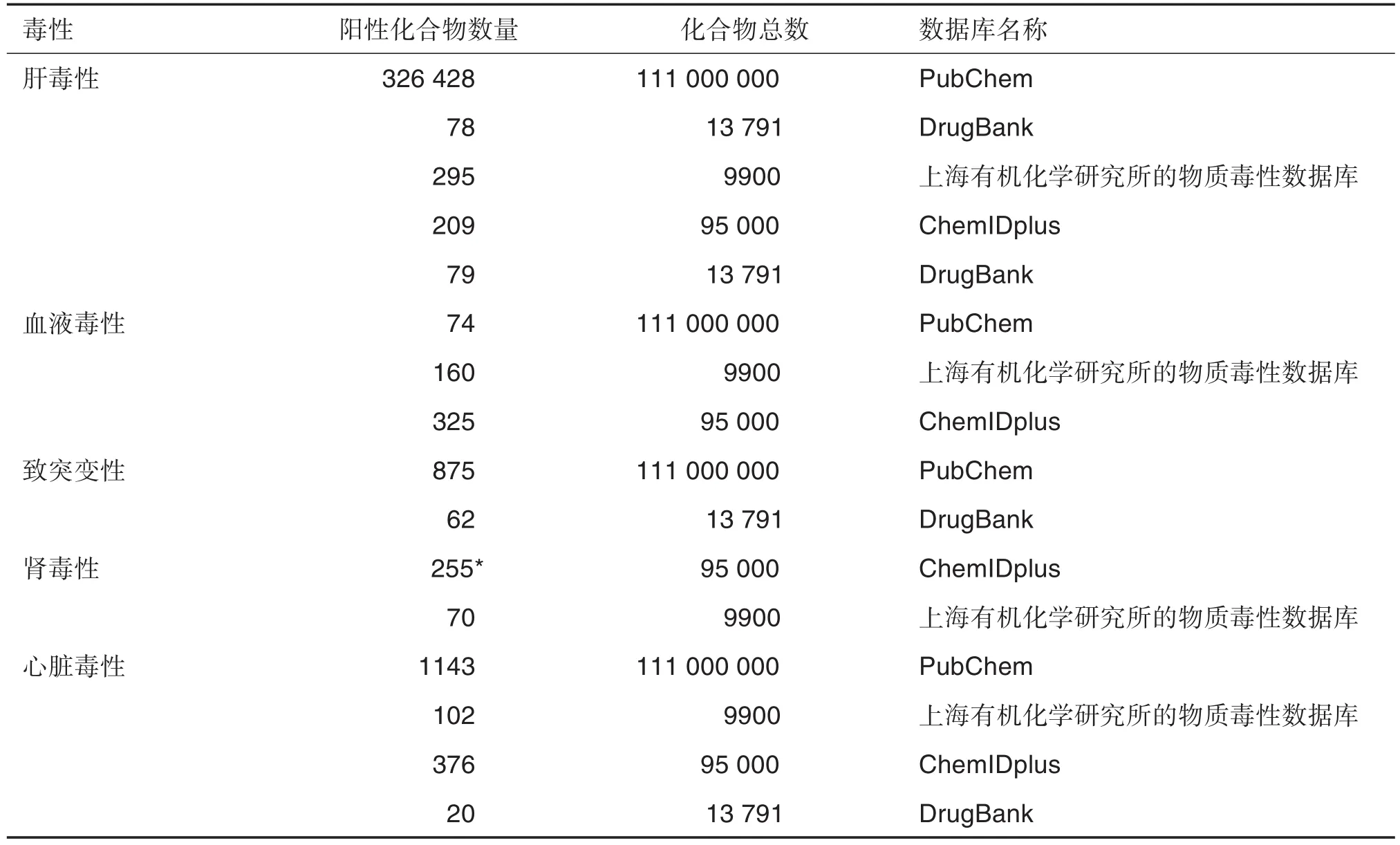
表1 常用毒性化合物数据库
1.2.2 深度学习模型的构建
在模型的构建过程中,一般采用分子描述符将化合物的结构和性质编码为模型能够直接识读和处理的数字化信息[25]。目前应用于计算机预测的分子描述符数量达到几百甚至上千,然而并无一种分子描述符普遍适用于编码所有化学信息学数据[26]。因此,应根据待解决的毒性预测问题和算法选择合适的分子描述符。
分子指纹是一种特殊的分子描述符,常见的有扩展联通指纹、PubChem分子库和分子接入系统等[27-29](表2)。分子指纹能将每一个分子的化学结构简化成独特的字串符,用于表征内部子结构或官能团是否存在,便于进行2个相似化合物的比较[30]。

表2 常用分子指纹
当前用于药物毒性识别与预测的DL模型大体可以分为2类:深度前馈神经网络模型和深度图神经网络模型。在深度前馈神经网络模型中,常用的深度神经网络模型是卷积神经网络(convolutional neural network,CNN)模型和循环或递归神经网络(recursive or recurrent neural network,RNN)模型。CNN模型是基于图卷积的分子编码方式,将输入的原子水平信息组合为分子水平的信息,尤其擅长由低到高层次的学习,从中提取出与任务最相关的一组特征[31]。在药物设计与研发领域,常使用的CNN模型有基于神经网络的分子指纹模型、基于分子图卷积(molecular graph convolution,MGC)模型和基于3D结构的MGC模型,目前已被用于预测分子性质和分子间的相互作用[32]。RNN是另一种前馈神经网络,其单元之间的连接能够形成环,该特点使RNN模型能够捕获并反映动态中的短暂行为。RNN模型在医学影像学中广泛用于辅助诊断,近年来也被用于药动学和药效学的相关研究[33-34]。无向图递归神经网络(undirected graph RNN,UGRNN)是RNN的一个新拓展,该方法将分子看作原子和键组成的无向图,依次以每个重原子为根节点,将其他节点的信息沿最短路径向根节点传递,每个根节点生成一个定长的向量,将所有定长向量相加得到分子水平的表征向量,输入全连接网络进行决策[35]。UGRNN模型减少了对分子描述符的依赖,在自动学习中即可得到合适的数据表示,已应用于预测化合物水溶性和药物分子肝毒性,并获得较高的准确率[36]。
与前馈神经网络将分子描述符作为输入不同,图神经网络(graph neural network,GNN)模型直接基于图对分子进行编码,其中分子中的原子作为图的节点,原子与原子之间的键作为边。GNN模型利用图卷积操作实现信息在节点间的传递、可提取基团等重要信息,在药物毒性识别和可解释性方面GNN模型具有明显优势。另外,GNN又可与CNN、RNN和注意力机制等联合使用形成多种深度GNN模型,如图注意网络模型、图卷积网络模型[37]和多任务图注意模型[38]等,这些模型在药物毒性识别中取得当前最好的结果。GNN模型、CNN模型和RNN模型的优缺点分析见表3。

表3 图神经网络、卷积神经网络和循环神经网络的优缺点
1.2.3 深度学习模型的验证与评估
模型生成后,下一步需对模型的拟合能力、稳定性以及预测能力进行验证与评价。验证包括内部验证和外部验证,分别用于评价模型的稳定性和模型的预测能力[37]。内部验证通过多次迭代和对测试集上的性能进行平均,可以减小随机分割数据集带来的偶然因素的影响,常见的方法是蒙特卡罗交叉验证[39]和 k折交叉验证[40]。外部验证使用独立于训练集的数据测试模型的泛化性,并检查是否出现“过拟合”问题[41]。
在模型性能评价中,常使用的指标包括灵敏性(sensitivity,SE)、特异性(specificity,SP)、准确性(accuracy,ACC)、精确率(precision,P)、ROC曲线下面积(area under the receiver operating characteristic curve,AUC)和 PR 曲线下面积(area under the precision recall curve,AUPR)[42]。在DL模型对药物毒性的预测结果中,SE即真阳性率,指所有毒性化合物中被正确预测为阳性的比例;SP即真阴性率,指所有无毒化合物中被正确地预测为阴性的比例;ACC即被正确预测为有毒的阳性化合物和被正确预测为无毒的阴性化合物占所有化合物的比例,准确率越高,模型越好;P为真毒性化合物数量占阳性结果数量的比例。
另外,ROC曲线是以假阳性率为横轴,SE为纵轴绘制的曲线。AUC值指随机给定一个有毒性化合物和一个无毒性化合物,模型输出有毒性化合物为阳性的概率值比输出无毒性化合物为阴性的概率值大的可能性。当AUC=0.5时,真阳性率和假阳性率相等,相当于随机猜测,AUC趋于1,即ROC曲线越靠近左上方时,模型的预测性能越好。与ROC曲线不同的是,PR曲线是以SE为横轴,以P为纵轴绘制的曲线。PR曲线越靠近右上方,表示模型的预测性能越好。
2 深度学习模型在药物毒性预测和毒理学研究中的应用
基于化学结构与毒性的关系,一般是使用已知的阳性化合物和阴性化合物的结构信息作为数据集进行模型训练和验证,使模型能够识别毒性化合物的结构特征。目前,DL模型已作为药物毒理学研究的工具,被用于解决药物分子及其代谢产物的毒性预测问题。然而,在药物毒理学研究中,除筛查出更多有潜在危害性的药物分子,还需进一步明确其致毒的生物学靶标和机制。因此,最近研发的DL模型同时把化学结构信息和生物学数据输入到模型中,使模型通过深度挖掘毒性药物分子的化学生物学数据以发现新的规律[43]。
2.1 预测药物分子的毒性
Xu等[36]使用NCTR数据集、Greene数据集、Xu数据集以及一个混合数据集的2788个化合物数据作为训练集,利用UGRNN分子编码方法构建药物肝损伤预测模型。在外部验证中,该模型的各性能指标均高于浅层神经网络架构的模型。2018年,Ambe等[44]针对肝细胞增生,在ORAD数据集、HESS数据集和一个混合数据集中比较3种机器学习算法,DNN,SVM,与RF预测化合物肝毒性的表现。经过验证,使用数据量较小的HESS数据集和SVM算法构建的模型获得最佳的预测性能。由此可见,在实际应用中,DL模型并不总是优于传统机器学习模型,这可能与数据集的大小有关。一般而言,传统机器学习模型适合处理规模较小的数据集,DL模型则在分析多且复杂的数据中显示出杰出的优势[45]。
类似地,基于化学结构的方法,Hua等[20]针对药物分子的血液毒性,使用从SIDER数据库、ChemIDplus数据库以及文献数据中筛选出的632个阳性化合物和1525个阴性化合物作为数据集,在未采用分子描述符的情况下直接将整个分子输入算法中,构建了预测药物分子血液毒性的CNN模型。在外部验证中,该模型ACC为76%,AUC为64%,SP为81%。由此可见,相较于传统机器学习模型,DL模型的另一个优势在于不依赖分子描述符编码,能将原始的输入信息进行转换。
2021年,Kumar等[46]使用从现有文献中筛选出的4053种化合物作为数据集,比较DNN,SVM,RF和k-近邻几种算法建立的模型在预测药物分子致突变性中的表现。通过测试,DNN模型在测试集和验证集中均获得最高的ACC,分别为95.0%和83.8%。表4总结了近年来用于药物毒性预测的DL模型。根据模型在外部验证集的ACC和AUC针对不同的药物毒性预测问题,DL模型均具有较高的预测能力。
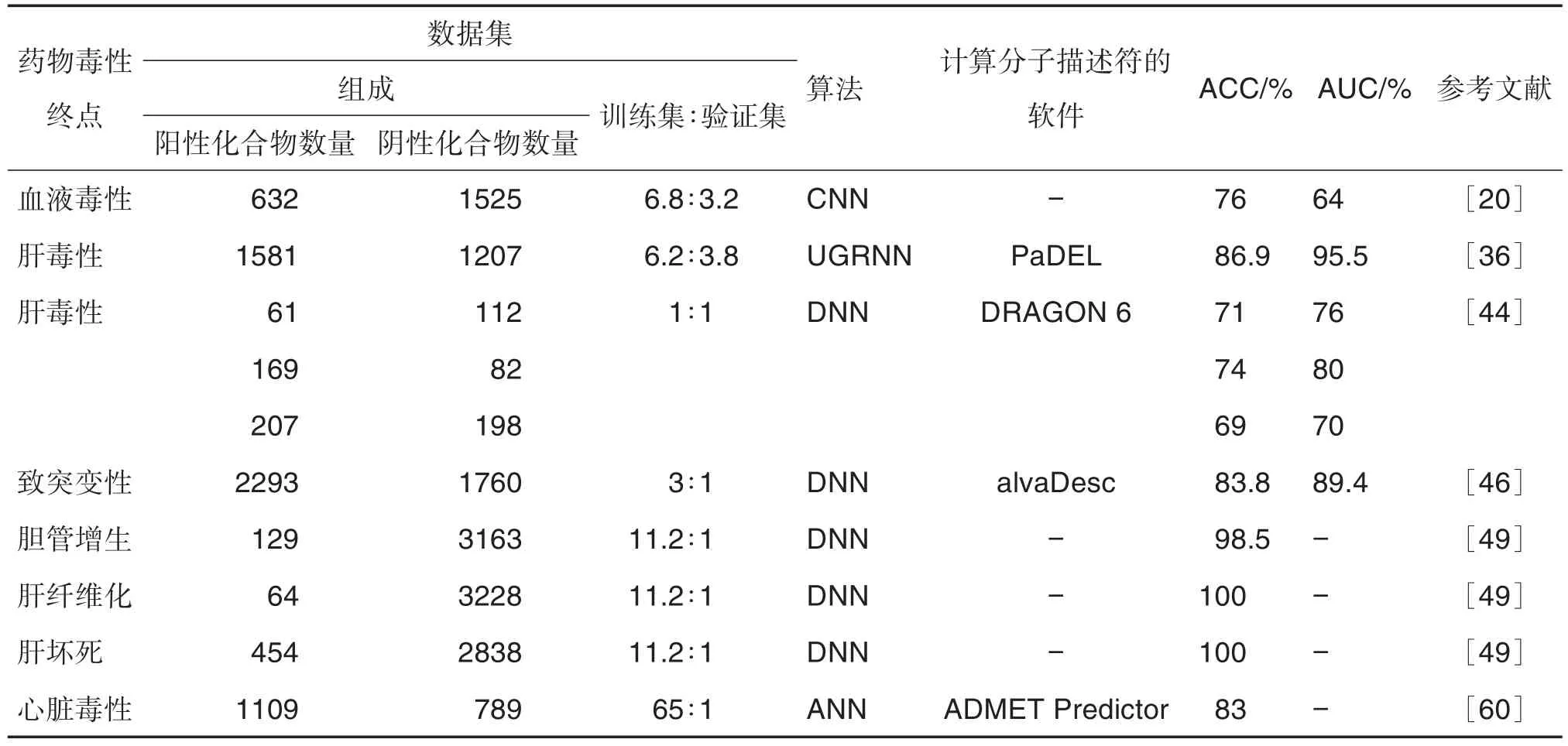
表4 深度学习模型预测药物不同毒性终点的性能
2.2 预测毒性反应性代谢产物的形成
2017年,Hughes等[47]首次构建出预测化合物经代谢活化形成醌类RM的CNN模型。他们在CNN架构中设置了1个输入层,3个隐藏层及3个输出层。隐藏层从低到高分别为原子层、原子对层、分子层,代表了药物分子在3个水平上形成醌类RM的能力。该CNN模型通过在0~1的范围内给每一层打分,以识别出化合物最有可能发生代谢活化的位点,并且综合评估化合物分子形成醌类RM的能力。在最近的一项研究中,Wang等[48]使用DL算法构建出预测药物分子的代谢反应类型及其潜在危害性代谢产物的机器学习模型。在药物毒理学的体内、体外研究中,DL模型能结合分子背景,较全面、准确、快速地预测出化合物经过转化形成毒性代谢产物而致毒的可能性,减少了实验的次数和周期。
2.3 预测药物分子致毒的生物学靶标
生物学数据集与DL模型结合的优势,一方面在于生物学数据涵盖从单个基因表达到多个基因富集通路上的分子水平信息,可以为模型提供成千上万的输入变量,DL模型通过增加隐藏层的数目能在同一时间对输入的变量进行学习,从而提高特征泛化能力;另一方面,在训练集中增加毒性相关基因和途径的生物学信息,利用DL模型的特征识别和提取能力,有助于发现药物分子致毒的生物学靶点和机制[43]。Wang等[49]针对胆管增生、肝纤维化和肝坏死3个肝毒性终端,使用DrugMatrix和OpenTG-GATEs数据库的9876个样本的毒理基因组学数据作为数据集,建立了预测肝毒性的DNN模型。该模型具有多个隐藏层,并且输入端和隐藏层均包含多个神经元,通过多次非线性转换可学习到更加复杂和抽象的特征。结果显示,在3种毒性终点基因和相关通路的识别中,与SVM和RF相比,采用DNN建立的模型显示出更好的稳定性和特异性。
3 深度学习模型在中药毒性预警中的挑战和展望
DL模型将有可能成为解决中药毒性预测这一难题的最优选择。目前,DL模型在中药研发领域方兴未艾,但同时面临一些瓶颈问题。
3.1 中药单体的毒性数据较少
DL模型要在中药成分的毒理学研究中发挥巨大效用,建立高质量的数据集是首要前提[50-51]。有研究表明,与仅由西药化合物构成的数据集训练出的模型相比,中药成分参与构成的混合数据库完善了训练集的化学结构空间,并有效降低机器学习模型的预测偏好性,提高了结果的准确性,因而更适用于中药成分的安全性评价[52]。然而,目前对于中药中具有潜在毒性的单体成分的研究相对较少,在国内外公共药物数据库中,中药单体的毒理学数据更是寥寥无几[53]。用于训练的毒理学数据有限,有可能使得DL模型在学习过程中泛化不全面,所学习到的毒性团仅限于数据集中的化合物。另外,通过文献检索得到的中药单体数据质量往往参差不齐。很多文献报道的因服用中药而引发的临床毒性事件中,无法排除医生辨证用药正确与否、患者是否误用滥用以及有无基础疾病等因素的影响,因此难以对检索到的中药单体进行正确的标记与分类[54]。
为获得高质量的数据集,建模前需要毒理学专家结合临床使用剂量和机体的复杂性进行全面分析,对数据进行严格的筛选、标注和分类。最根本的是要重视具有潜在毒性的中药单体“结构预警子”方面的深入研究,不断积累相关的实验数据,从源头上解决中药单体毒理学数据较少的问题。
3.2 深度学习模型的黑箱问题
由于DL架构过程是端到端,由输入端到输出端的因果关系的可解释性是DL模型落地于中药毒性预测必需的“通行证”[55-56]。为此,可以从2方面完善DL模型的可解释性。一方面,从预测原理方面入手,结合“结构预警子-毒性”关系和生物学致毒机制解释模型的决策依据,提高预测结果的可信度;另一方面,从模型的架构入手,通过调试函数对模型的架构造成微小的扰动,以确定不同变量对模型的影响,便于理解模型的行为和改进模型[57-58]。
3.3 深度学习模型在中药毒性预警中的展望
借助于集成式建模软件,如ADMET Predictor和Virtual Tox Lab,机器学习模型已投入到中药毒理学研究并展现出良好的预测性能[59-60]。如李雅秋等[60]针对心脏毒性使用ToxRefDB数据库和SIDER数据库中1109个阳性化合物和789个阴性化合物作为训练集,另外使用29种已知毒性的中药成分组成外部验证集,并采用ADMET Predictor软件中自带的SVM和ANN算法模块构建模型。在外部验证集中,ANN模型预测的SE为95%,SP为60%,ACC为83%,明显优于SVM模型。
He等[61]使用由检索科学出版物得到的296个肝毒性中药成分和584个无肝毒性中药成分以及公开的CTD数据库组成的数据集,利用多种传统机器学习算法和分子描述符构建了数个基本分类器,将其中预测性能最佳的基本分类器进行组合,建立了一款预测中药肝毒性的模型。经检验,该组合型预测模型在交叉验证和外部验证中均表现出优于单个分类器的预测性能。值得一提的是,虽然组合式学习能够通过增加基本分类器的数目提高传统机器学习算法处理复杂数据的能力,但各基本分类器的预测性能不一,组合后不一定能获得优势互补的效果。迄今为止,DL模型在中药成分毒性评价中的应用还比较少。
总体来看,传统机器学习模型高度依赖人工设计的输入特征,普适性低且易出现“过拟合”;DL模型处理分析大数据和复杂数据更胜一筹。针对中药多组分、多靶标的特点,DL模型有能力深度挖掘出中药毒性成分的结构和生物学特征。若在中药研发早期利用DL模型实现高效、快速的风险预警,既能把毒性大的中药成分及时剔除,也能为具有潜在危害性的药物候选分子的结构优化提供理论指导[20]。
4 结语
中药成分复杂,筛查具有潜在毒性的中药成分并揭示其致毒机制是当前亟待解决的难题。在药物的毒性评价中,与耗损率高、周期长的实验室方法相比,DL模型几乎不需要成本、人力和物力,即可快速、准确地给出预测结果。与传统机器学习模型相比,DL模型在处理和分析复杂体系的信息上更具优势。虽然目前在数据来源和模型可解释性方面仍有不足,但DL模型在中药毒性预警中的广阔应用前景不可否认。随着DL模型的不断优化以及更多潜在毒性中药成分被揭示和积累成规范的数据库,相信在不久后,DL模型将突破现有的瓶颈,成为新一代预测中药潜在毒性的杰出工具。
猜你喜欢
四川蚕业(2022年2期)2022-11-19
当代水产(2022年6期)2022-06-29
包装工程(2022年1期)2022-01-26
当代水产(2021年6期)2021-08-13
意林原创版(2021年7期)2021-08-03
昆明医科大学学报(2021年2期)2021-03-29
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
