
气相色谱法测定黄精中甲胺磷残留量的不确定度评定
2022-04-11 11:54金雅慧俞华芬李罗飞
现代农药 2022年2期
金雅慧,俞华芬,李罗飞,秦 丽*
(1.杭州市余杭区农产品质量安全检验检测站,杭州 311100;2.宁波工程学院,奉化研究院,浙江宁波 315500)
黄精(Polygonatirhizoma)是百合科黄精属植物滇黄精、黄精、多花黄精的干燥植物根茎[1]。现代药理学研究发现,黄精的化学成分主要有多糖类、甾体皂苷、三萜皂苷类、黄酮类和蒽醌类、木脂素类、植物甾醇类、氨基酸以及金属元素类等,其中黄精多糖为主要药效成分[2-4]。《中华人民共和国药典》(2020年版)在通则0212《药材和饮片检定通则》中增订了针对控制农药残留污染的内容,要求药材和饮片(植物类)中不得检出33种禁用农药[5]。甲胺磷(Methamidophos),化学式为C2H8NO2PS,属于广谱缓释有机磷类杀虫剂,具有良好的内吸、胃毒和触杀作用,广泛用于各种农作物病虫害防治[6],是通则0212中规定的不得检出的禁用农药之一。
测量不确定度是一种对测量/检测结果可疑程度的定量表述方法,能反映检测人员、测量仪器设备和检测实验室环境条件等的测量能力、水平。我国GB/T 27025—2019《检测和校准实验室能力的通用要求》[7]和RB/T 214—2017《检验检测机构资质认定能力评价检验检测机构通用要求》[8]中规定:检测和校准实验室都必须进行测量不确定度的评定,且应在具体的检测和校准工作中应用这些程序来进行测量不确定度的评定,并能运用测量不确定度评定的结果来判定检测结果是否符合规定要求以及对检测数据或测量过程实施监控。目前,已有相关文献[9-12]采用农业行业标准NY/T 761—2008《蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留的测定》[13]测定蔬菜、水果中有机磷农药残留量的不确定度,如采用气相色谱测定鲜枸杞中农药氧乐果残留的不确定度评定[14];采用气相色谱-串联质谱测定三七中农药腐霉利含量的不确定度评定[15]等,而采用《中国药典》(2020年版)通则2341《农药残留测定法》[16]测定黄精中甲胺磷残留量的不确定度评定未见报道。本文按照《中国药典》通则2341《农药残留测定法》中《第二法有机磷农药残留量测定法(色谱法)》[16],并参照JJF 1135—2005《化学分析测量不确定度评定》[17]及JJF 1059.1—2012《测量不确定度评定与表示》[18],对黄精中甲胺磷残留检测过程中各不确定度分量进行来源分析和计算评定,以期为提升检测结果的准确性和可靠性提供有效依据,同时为使用气相色谱测定黄精中其他有机磷农药残留量的不确定度评定提供参考。
1 材料与方法
1.1 材料与试剂
乙酸乙酯、无水硫酸钠、正己烷,以上均为分析纯,无锡市晶科化工有限公司;滤膜(0.22μm),北京莱博润科生物科技有限公司;石墨化炭黑固相萃取SPE小柱(250 mg/3 mL),广州太玮生物科技有限公司;甲胺磷标准品1 000μg/mL(纯度99.9%),农业部环境保护科研监测所。
1.2 仪器与设备
K-600型商用食品加工器,德国博朗公司;PL 202-L型电子天平,梅特勒-托利多国际贸易有限公司;S10200-A型超声波清洗器,天津奥特塞恩斯公司;AR-200型旋转蒸发仪,瑞士布奇公司;MS-3漩涡混合器,德国艾卡公司;N-EVAP 12位氮吹仪,美国Organomation公司;Agilent 7890A型气相色谱仪(FPD检测器),美国安捷伦公司。
1.3 试验方法
1.3.1 标准溶液配制
准确移取0.5 mL甲胺磷标准品1 000μg/mL,用乙酸乙酯定容于50 mL玻璃容量瓶中,得到甲胺磷标准储备液10.0 mg/L。准确移取0.5 mL上述甲胺磷标准储备液,用乙酸乙酯定容于50 mL玻璃容量瓶中,得到0.1 mg/L甲胺磷标准溶液,再分别移取0.5、1.0、2.0、5.0 mL上述甲胺磷标准储备液,用乙酸乙酯定容于10 mL玻璃容量瓶中,得到0.5、1.0、2.0、5.0 mg/L甲胺磷标准溶液。甲胺磷标准曲线系列质量浓度为0.1、0.5、1.0、2.0、5.0 mg/L。
1.3.2 气相色谱条件
色谱柱:HP-5(30 mm×0.32 mm×0.25μm);载气:N2(纯度99.99%);进样口温度:220℃;进样模式:不分流进样;进样体积:1.0μL;恒流模式:流速3.53 mL/min;检测器温度:250℃;尾吹60 mL/min,燃气75 mL/min,助燃气100 mL/min;程序升温:60℃(保持2 min),5℃/min升温至250℃(保持2 min)。
1.3.3 样品前处理
采用《中国药典》(2020年版)通则2341中第二法的前处理方法[16],称取粉末黄精试样5 g,加入无水硫酸钠5 g、乙酸乙酯50 mL,于冰浴中超声3 min,静置15 min后收集上层滤液,加入乙酸乙酯30 mL至固体部分,于冰浴中超声2 min,静置15 min后收集上层滤液;合并上述上层滤液,使用少量乙酸乙酯冲洗滤纸及固体部分,并入上述滤液。将滤液于40℃以下浓缩近干,转移至5 mL玻璃试管中,使用乙酸乙酯稀释至5 mL;吸取上述溶液1 mL,上样至石墨化炭黑固相萃取SPE小柱(乙酸乙酯5 mL预淋);使用正己烷-乙酸乙酯(1∶1,V/V)混合液5 mL进行洗脱;收集洗脱液,氮吹浓缩近干,用乙酸乙酯定容至1 mL,涡旋混匀后,过0.22μm有机滤膜至进样瓶,供气相色谱仪测定。
1.3.4 数学模型的建立
构建的数学模型为乘除形式,可用相对不确定度评定,无需计算灵敏度系数。样品中被测农药甲胺磷的残留量按式(1)计算。
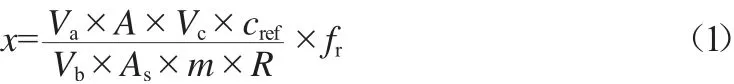
式中:x为黄精中甲胺磷的残留量,mg/kg;Va为提取溶剂(乙酸乙酯)的总体积,mL;Vb为用于检测的提取溶液的体积,mL;Vc为样品溶液处理后定容的体积,mL;cref为标准溶液中甲胺磷质量浓度,μg/mL;A、As分别为样品溶液和标准溶液中甲胺磷峰面积的平均值,mAU·s;m为样品的质量,g;R为样品加标回收率,%;fr为样品均匀性校正因子,取值为1。
黄精中甲胺磷残留量相对标准不确定度的合成按式(2)计算。

式中:urel(rep)为检测过程总重复性产生的相对不确定度,是由气相色谱仪峰面积A、As、称量、移取溶液、定容等操作的重复性分量合并而成的一个总检测过程中的分量,利用X重复测量数据计算的标准偏差;urel(crep)为甲胺磷标准溶液配制过程中产生的相对不确定度;urel(sv)为甲胺磷标准曲线拟合产生的相对不确定度;urel(m)为黄精样品称量产生的相对不确定度;urel(Va)、urel(Vb)、urel(Vc)分别为样品提取液、用于检测的提取液、样品溶液处理后定容产生的相对不确定度;urel(R)为方法回收率产生的相对不确定度;urel(fr)为黄精样品的均匀性校正因子fr产生的相对不确定度;urel(A)为气相色谱峰面积测量值产生的相对不确定度。
2 结果与分析
2.1 测量不确定度来源分析
从数学模型、前处理过程、检测原理等方面分析,用气相色谱法测定黄精中甲胺磷残留量的不确定度来源,如图1所示。

图1 不确定度来源
2.2 不确定度分量评定
2.2.1 检测过程总重复性产生的相对不确定度urel(rep)
分别称取10份黄精样品,按1.3试验方法进行测定残留量x,按贝塞尔公式,即式(3)计算x的相对标准偏差s(x),重复测定检测结果见表1。
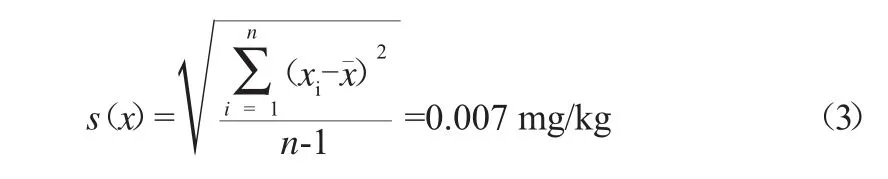

表1 重复测定检测结果(n=10)
实际测量时,一般将对样品测定2次作为测量结果,则检测过程总重复性产生的相对不确定度urel(rep)按式(4)计算。

2.2.2 甲胺磷标准溶液配制过程中产生的相对不确定度urel(crep)
2.2.2.1 甲胺磷标准品浓度cs产生的相对不确定度urel(cs)
使用有证标准品,甲胺磷质量浓度标准值为1 000μg/mL,标准品证书记录甲胺磷不确定度为3%,即±30μg/mL(k=2),甲胺磷标准品产生的相对标准不确定度为
2.2.2.2 甲胺磷标准溶液稀释配制过程中由玻璃量器、液体体积膨胀产生的相对不确定度urel(Vi)(i=1~5)
根据JJG 196—2006《常用玻璃量器检定规程》[19]规定:1、2、5 mL玻璃分度移液管(A级)的最大允许误差(MPE)分别为±0.008、±0.012、±0.025 mL,按照均匀分布处理玻璃单标线容量瓶的MPE值分别为±0.020、±0.050 mL,按照三角分布处理查询本实验室当日环境控制记录,实验室环境温度在(20±5)℃,与常用玻璃量器的检定校准温度(20℃)[19]不同,温度变化产生的不确定度可通过估算温度变化范围,体积膨胀系数来计算。因常用有机溶剂的体积膨胀系数远高于常用玻璃量器的体积膨胀系数,故只须考虑常用有机溶剂的体积膨胀。乙酸乙酯体积膨胀系数为1.38×10-3/℃,按照均匀分布处理
使用分度移液管、单标线容量瓶等稀释配制甲胺磷标准溶液过程中,有2个主要不确定度来源:最大允许误差和温度影响,且这2个分量互不相关,其合成标准不确定度可以采用方和根方法合成,其相对标准不确定度可采用合成标准不确定度除以所量取体积进行估算。结果如下:

2.2.2.3 甲胺磷标准溶液配制过程中产生的相对不确定度urel(crep)
在甲胺磷标准溶液配制过程中,使用1 mL分度移液管(A级)吸取0.5 mL(V1-0.5)液体2次,吸取1 mL(V1-1)液体1次;使用A级2、5 mL分度移液管吸取2 mL(V2)液体1次、5 mL(V3)液体1次;使用10 mL(V4)、50 mL(V5)单标线容量瓶次数分别为4、1次,合成甲胺磷标准溶液配制过程中产生的相对标准不确定度urel(crep)按式(5)计算。

2.2.3 甲胺磷标准曲线拟合产生的相对不确定度urel(sv)
对质量浓度为0.1、0.5、1.0、2.0、5.0 mg/L的甲胺磷标准溶液进行测定,以甲胺磷质量浓度c为横坐标,气相色谱测得的峰面积y为纵坐标,采用最小二乘法进行线性拟合,得到拟合直线方程y=ac+b为y=5 037.1c+57.954(R2=0.999 9)(表2)。

表2 标准曲线校正引入的相对不确定度
标准溶液峰面积的余差标准偏差Sy按式(6)计算。

标准曲线拟合引入的相对不确定度urel(sv)按式(7)计算。
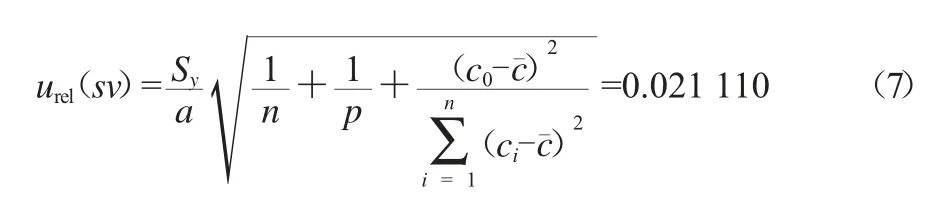
式中:c0为被测样品质量浓度的平均值,取值为0.213 mg/L;cˉ为甲胺磷标准溶液中各质量浓度的平均值,取值为1.72 mg/L;ci为甲胺磷标准溶液中的一系列质量浓度值,mg/L;a为拟合直线方程中的斜率,取值为5 037.1;b为直线截距,取值为57.954;n为作拟合直线方程时甲胺磷标准溶液的测定次数(每个浓度测定2次),取值为10;p为黄精样品测定次数,取值为2。
2.2.4 样品称量产生的相对不确定度urel(m)
用电子天平称量黄精样品,电子天平检定合格。经查询该天平MPE值为±0.05 g,按均匀分布处理,称量采用去皮法,则样品称量产生的相对不确定度urel(m)按式(8)计算。
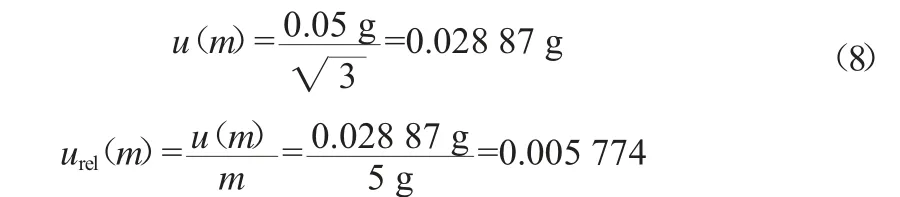
2.2.5 样品提取液、用于检测的提取液、样品溶液处理后定容产生的相对不确定度urel(Va)、urel(Vb)、urel(Vc)
根据JJG 196—2006《常用玻璃器检定规程》[19]规定:5 mL刻度试管、1 mL分度移液管(A级)的MPE值分别为±0.100、±0.008 mL,按照均匀分布处理,实验室温度在(20±5)℃,乙酸乙酯体积膨胀系数为1.38×10-3/℃,按照均匀分布处理
使用分度移液管吸取样品提取液、用于检测的提取液、样品溶液处理后定容等过程中,有2个主要不确定度来源:最大允许误差和温度影响,且这2个分量互不相关,其合成标准不确定度可以采用方和根方法合成,其相对标准不确定度可采用合成标准不确定度除以所量取体积进行估算。结果如下:

式中:因样品溶液处理后是在5 mL刻度试管中定容至1 mL,故MPE取值为0.100,并以此计算urel(Vc)。
2.2.6 方法回收率产生的相对不确定度urel(R)
使用5份阴性样品做加标试验,甲胺磷添加浓度为0.010 mg/kg,得回收率(R)分别为102.2%、101.4%、99.0%、100.6%、99.8%,平均回收率(R)为100.6%。
采用A类不确定定评定中的极差法进行评定回收率产生的相对不确定度urel(R)。查询JJF 1059.1—2012《测量不确定度评定与表示》[18]中极差系数及自由度表可知,当测量次数n=5时,极差系数C=2.33,按式(9)计算urel(R)。
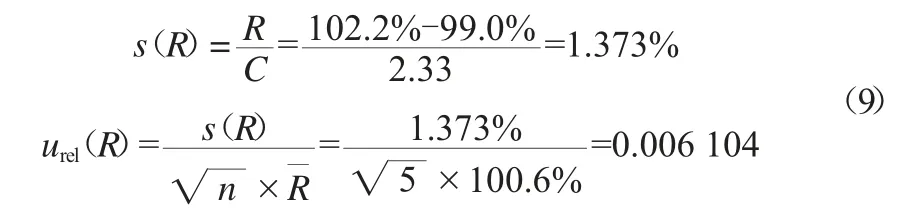
通过t检验,判断回收率(R)是否与100%存在显著差异。查询t值表,在置信率P=95%,t0.95(4)=2.780,按式(10)计算检验值t。

由此说明,回收率(R)不必在测量模型中采用修正测定结果,但其对不确定度的贡献需考虑。
2.2.7 样品的均匀性校正因子(fr)产生的相对不确定度urel(fr)
根据本文所采用的检测方法,平行配制3份黄精样品,在气相色谱仪上进行测定,得到测定数据x′为0.506、0.510、0.503 mg/kg,xˉ′为0.506 mg/kg。
采用A类不确定度评定中的极差法进行评定。查询JJF 1059.1—2012《测量不确定度评定与表示》[18]中极差系数及自由度表可知,当测量次数n=3时,极差系数C=1.69。
因样品测定以2次测量平均值为其测量结果,则样品的均匀性校正因子产生的相对不确定度urel(fr)按式(11)计算。

2.2.8 气相色谱峰面积测量值产生的相对不确定度urel(A)
2.2.8.1 气相色谱进样体积产生的相对不确定度urel(Ve)
由1.3.2气相色谱条件可知,气相色谱进样体积为1.0μL,查询气相色谱仪进样微量注射器说明书,其定量重复性为1.0%,即其重复性相对标准不确定度为
2.2.8.2 气相色谱FPD检测器产生的相对不确定度urel(A)1
查询气相色谱检定证书可知,定量重复性为1.3%,按照矩形分布处理采用B类不确定度评定,则气相色谱FPD检测器产生的相对标准不确定度为
2.2.8.3 气相色谱峰面积响应值产生的相对不确定度urel(A)2
仪器对在相同色谱条件下所测样品和甲胺磷标准溶液的影响是一致的,因此气相色谱峰面积响应值产生的相对不确定度urel(A)2可忽略不计,气相色谱峰面积的重复性已在检测过程总重复性中纳入考虑。
2.2.8.4 气相色谱峰面积测量值产生的相对不确定度urel(A)
峰面积测量值产生的相对不确定度urel(A)按式(12)计算。

2.3 合成标准不确定度
上述各项相对标准不确定度分量统计结果见表3,通过各项相对标准不确定度合成得到黄精样品中甲胺磷残留量的合成相对标准不确定度为:

表3 相对标准不确定度分量统计结果

黄精样品中甲胺磷残留量在实际2次测量的平均值x为0.213 mg/kg,则合成标准不确定度按式(13)计算。
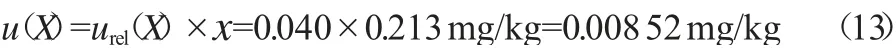
2.4 扩展不确定度U(X)和测量结果表示
根据JJF 1135—2005《化学分析测量不确定度评定》[17],在95%置信概率下,包含因子k=2,则扩展不确定度按式(14)计算。
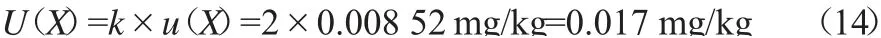
当测得黄精中甲胺磷残留量为0.213 mg/kg时,其测量结果表示为0.213±0.017 mg/kg,k=2。
3 结 论
本研究采用气相色谱法测定黄精中甲胺磷残留量的不确定度,并对整个定量检测过程中可能产生的各种不确定度分量进行来源分析、计算评定。由分析结果可知,甲胺磷标准曲线拟合、甲胺磷标准溶液配制的不确定度分量相对贡献较大,黄精样品称量、前处理过程中各所涉液体体积、方法的回收率、样品的均匀性校正因子等不确定度分量贡献相对较小。因此,在进行农药残留定量检测过程中,可通过判断选择合适的玻璃量具,并定期做好各玻璃量具的检定和校准工作。同时,应控制好检测实验室的环境条件,以及通过质量监督、能力验证、人员比对等质量管理措施,不断提高实验操作人员的操作能力水平,以期达到降低检测结果测量不确定度的目的,确保检测结果的准确、可靠。
猜你喜欢
新农业(2022年16期)2022-11-07
食品安全导刊·中旬刊(2022年3期)2022-04-15
甘肃农业科技(2020年6期)2020-07-16
科技创新导报(2020年5期)2020-06-11
婚育与健康(2020年12期)2020-02-26
分析化学(2019年3期)2019-03-30
科教导刊(2017年26期)2017-11-07
科技与创新(2015年17期)2015-09-11
家庭医药·快乐养生(2015年8期)2015-09-10
科技与创新(2014年12期)2014-08-28
