
超高效液相色谱法测定噁草酸原药及乳油中有效成分含量
2022-04-06 06:15王皙玮
黑龙江大学工程学报 2022年1期
刘 琳,周 芹,2,3,*,王皙玮,2,3
(1.黑龙江大学 现代农业与生态环境学院,哈尔滨 150080;2.农业农村部甜菜品质监督检验测试中心,哈尔滨 150080;3.农业农村部糖料产品质量安全风险评估实验室,哈尔滨 150080)
0 引 言
噁草酸又名喔草酯(propaquizafop),化学名称为:2-异亚丙基氨基氧乙基(R)-2-(4-(6-氯喹喔啉-2-基氧)苯氧基)丙酸酯,化学分子式为C22H22CLN3O5,其结构式见图1,属于芳氧苯氧丙酸酯类除草剂[1],主要用于防除一年生和多年生禾本科杂草[2]。为乙酰辅酶A羧化酶(ACCase)抑制剂,通过阻碍ACCase来抑制脂肪酸的合成,干扰细胞膜的形成,从而使杂草生长停止,最终死亡,发挥除草的功能。具有内吸性,对大豆、棉花、油菜、马铃薯和蔬菜等安全,在杂草幼苗期和生长期施药效果最好,且作用迅速[3]。

图1 噁草酸的结构式
自从上世纪80年代第一例芳氧苯氧丙酸酯类除草剂禾草灵被开发,就以其高效、低毒、高选择性的优良特点,受到广泛关注[4]。随后,不断合成出新品种,诸如苯并噻唑基、苯并咪唑基、喹啉基和吡啶并唑基等一系列新型芳氧苯氧丙酸酯类化合物,而到了90年代随着对现有除草剂作用机制研究的深入,出现了基于乙酰辅酶A羧化酶的分子设计研究[5],目前商品化的品种已达20多种,主要包括精喹禾灵、高效氟吡甲禾灵、噁草酸等,与其他的芳氧苯氧丙酸酯类除草剂相比,噁草酸主要用在大豆、棉花、油菜、甜菜、马铃薯、花生和蔬菜等作物上,2018年原药及10%乳油在我国首次获得登记。
目前,对于噁草酸的研究,主要集中在药效[6-8]及残留的检测。郭松年等[9]利用 QuEChERs-液质联用法测定菜籽油中7种芳氧苯氧丙酸酯类除草剂残留,其中就包括噁草酸,采用乙腈提取、分散固相萃取对样品进行前处理,质谱采用MRM监测模式,回收为78.3%~95.1%,具有基质效应低、处理简单、准确快速、重复性好的优点;Shin Y等[10]同样采用液相色谱-质谱联用的方法建立了噁草酸及352种农药在食用昆虫中的联合测定,方法具有较好的的准确度及精密度;吴春英等[3]同样采用液相色谱-质谱联用的方法测定了水中的16种芳氧苯氧丙酸酯类除草剂,通过优化前处理条件及仪器条件,建立的方法具有检出限低、回收率高的优点。Saito-Shida S等[11]利用液相色谱-四级杆-飞行时间质谱(UPLC-Q-TOF-MS),建立了粮谷及豆类中噁草酸等153种农药残留的测定方法,结果表明,LC-QTOF-MS方法适用于谷物和豆类中农药残留的常规分析。噁草酸结构中含有共轭双键,在紫外区具有光谱吸收,采用液相色谱分离,基质包括土壤、植物、水以及食用昆虫等。目前的研究都是针对农药残留的检测,制剂有效成分检测的报道较少。近年来,在我国随着农药使用数量的增多,农药产品质量参差不齐,根据《农药管理条例》规定,农药所含有效成分种类与农药的标签、说明书标注的有效成分不符,均视为假农药[12-13],所以对制剂有效成分含量的检测具有重要意义,本研究拟建立一种方法可以同时适用于噁草酸原药(Technical material,简写为TC)及乳油(Emulsifiable concentrate简写为EC)中有效成分的检测,采用超高效液相色谱法,通过波长扫描确定适宜的检测波长,特异性检验确定色谱峰纯度,优化了流动性比例,建立的方法具有灵敏度高,操作简便的特点,并为国家标准的制定提供依据。
1 材料与方法
1.1 实验材料及仪器
甲醇:色谱纯。水:新蒸二次蒸馏水或超纯水。噁草酸标样:已知质量分数为98.5%。Waters超高效液相色谱仪(UPLC),二极管阵列检测器(PDA),色谱柱:Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm)。过滤器:滤膜孔径约0.45 μm。超声波清洗器。
1.2 实验方法
色谱操作条件:流动相:Ψ(甲醇∶水)=80∶20,经滤膜过滤,并进行脱气。流速:0.2 mL·min-1。柱温:35 ℃±2 ℃。检测波长:235 nm。进样体积:0.5 μL。标样溶液和试样溶液的制备:称取0.05 g(精确至0.000 1 g)噁草酸标样于100 mL容量瓶中,加入20 mL甲醇,超声波振荡5 min,冷却至室温,用甲醇稀释至刻度,摇匀。测定及计算方法:在上述操作条件下,待仪器稳定后,连续注入数针标样溶液,直至相邻两针噁草酸峰面积相对变化<1.2%时,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。将测得的两针试样溶液以及试样前后两针标样溶液中噁草酸峰面积分别进行平均。计算试样中噁草酸的含量。
2 结果及分析
2.1 检测波长的选择
噁草酸分子中存在共轭双键,价电子会发生能级跃迁,进而产生紫外吸收光谱。为了找到噁草酸吸光值最大处吸收波长,利用UPLC-PDA在200~400 nm进行波长扫描,紫外光谱图见图2。由图2可见,噁草酸的最大吸收波长为235.2 nm,在农药分析中,一般情况下,如果最大吸收波长处无干扰,分析检测时选用最大吸收波长[14],本实验中235 nm波长处灵敏度较高,干扰小,能够满足分析的要求,故将检测波长确定为235 nm。
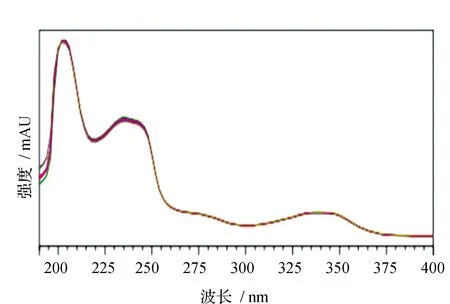
图2 噁草酸的紫外光谱图
2.2 色谱柱及流动相的选择
根据噁草酸的性质,选用反相色谱进行分析,常用的色谱柱包括C18、C8等,C18一般用来分析分子量较小的物质,C8适合分析大分子类的物质。由于C18比C8的碳链更长,带来更好的保留特性。本实验中选择常用的C18反相柱,根据噁草酸物化性质和溶剂的紫外吸收波长,选择甲醇作为溶剂溶解样品,并以甲醇和水作为流动相,将流动相按不同比例在色谱柱上进行试验,随着甲醇比例的增加,保留时间减小,最终选择Ψ(甲醇∶水)=80∶20作为流动相,流速0.2 mL·min-1。该条件下,噁草酸色谱峰峰形较好,与杂质能完全分离(图3),具有良好的精密度和准确度,并且分析时间较短,提高了工作效率。

图3 噁草酸原药及10%噁草酸乳油的UPLC色谱图
2.3 特异性实验
特异性就是对峰纯度进行检验,利用二极管阵列检测器全波长同时采集数据的特点,也赋予了其光谱比对的功能。峰纯度色谱图见图4。由图4可见不同的化合物具有不同的紫外吸收光谱形状和吸光特性,从而实现对色谱峰纯度的检测。采用UPLC-PDA峰纯度分析法来鉴别噁草酸。一般用纯度角度与纯度阈值比较,如果前者大于后者,说明峰纯度不够,有共流出现象,反之,则认为峰纯度符合要求。本实验中噁草酸标样、92%噁草酸原药、10%噁草酸乳油中的噁草酸峰纯度角度均小于纯度阈值,有效成分处无其它物质干扰,符合定量分析要求。
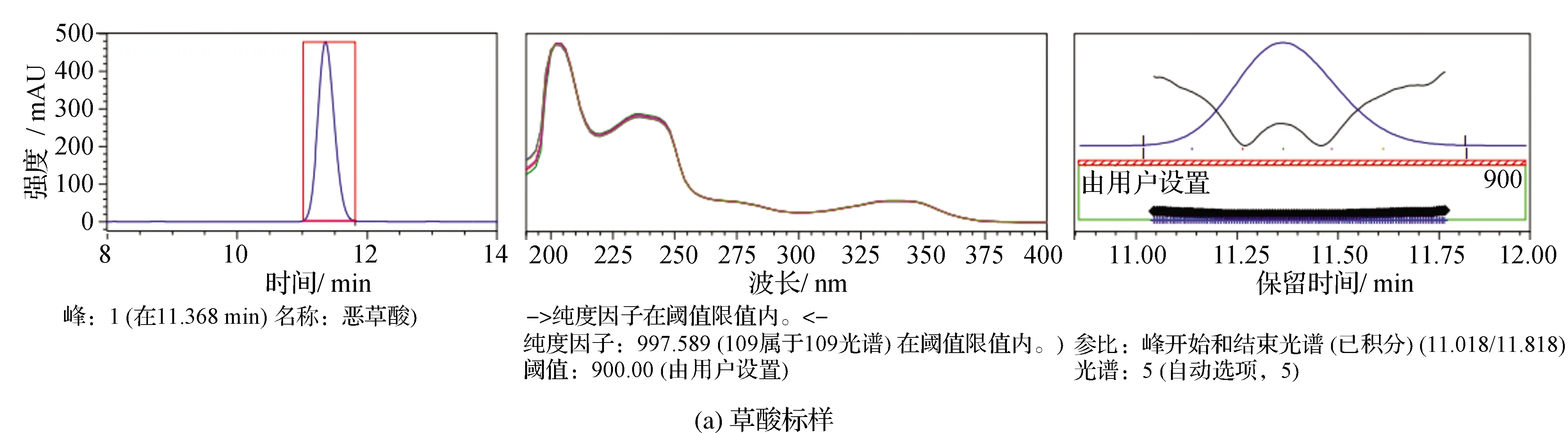
图4 HPLC-PDA峰纯度色谱图
2.4 方法学考察
2.4.1 线性关系
按标样溶液的制备方法配制5个不同浓度的有效成分线性相关溶液,分别标记为STD1至STD5。待仪器稳定后,按照STD1至STD5的顺序测定每个溶液中噁草酸的峰面积,取两次测定的平均结果。以噁草酸质量浓度为横坐标,峰面积为纵坐标绘制标准曲线见图5。由图5可见当噁草酸质量浓度为100.5~1 003.9 mg·L-1(进样体积1 μL),与相应的噁草酸峰面积之间呈现良好的线性关系,计算得回归方程为y=39 050x+97 636,相关系数R2=0.999 5,完全可以满足定量分析要求。本方法中噁草酸标样质量浓度为 502.4 mg·L-1。

图5 噁草酸标准曲线
2.4.2 方法精密度试验
按试样溶液的制备方法配制5个92%噁草酸原药精密度溶液,分别标记为TC-A1~TC-A5;5个10%噁草酸乳油精密度溶液,分别标记为EC-A1~EC-A5。以有效成分线性相关溶液STD3为标样溶液,待仪器基线稳定后,按照标样溶液、精密度溶液、精密度溶液、标样溶液的顺序进行测定,结果见表1~表2。农药测定和分析方法中,数据结果的确认应以修改的Horwitz公式(2(1-0.51ogC)×0.67)为依据[15],RSD值小于这个修正值通常被认为是合格的。由表1~表2可见,92%噁草酸原药、10%噁草酸乳油中噁草酸质量分数测定结果的RSD分别为0.39%、0.21%,小于修改的Horwitz公式2(1-0.51ogC)×0.67=1.36(其中C=0.905 0),表明有效成分分析方法精密度的测定结果符合要求。

表1 噁草酸原药精密度试验结果

表2 噁草酸乳油精密度试验结果
2.4.3 方法准确度试验
分别称取约含0.025 g(精确至0.000 1 g)噁草酸的92%原药、10%噁草酸乳油于100 mL容量瓶中,再加入噁草酸标样约0.025 g(精确至0.000 1 g),按2.5.2试样溶液的制备方法配制5个有效成分准确度溶液,分别标记为:TC-B1~TC-B5、EC-B1~EC-B5。以线性相关溶液STD3为标样溶液,在上述操作条件下,待仪器基线稳定后,按照标样溶液、准确度溶液、准确度溶液、标样溶液的顺序进行测定,结果见表3~表4。92%噁草酸原药、10%噁草酸乳油中噁草酸平均回收率分别为100.78%、100.44%,具有良好的准确度。
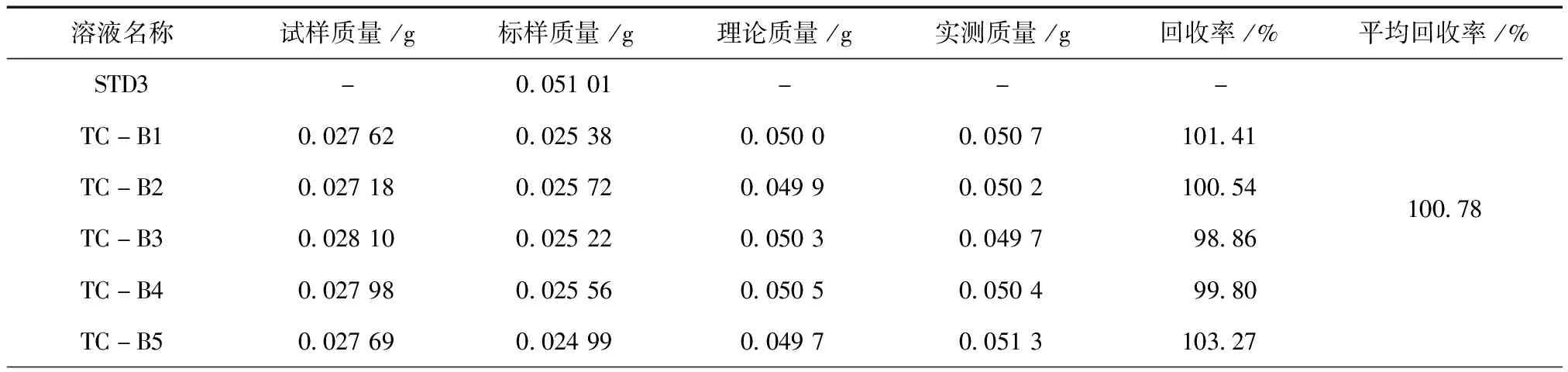
表3 噁草酸原药回收率实验结果

表4 噁草酸乳油回收率试验结果
2.4.4 不同实验室间方法验证
为了验证建立的方法在不同实验室的适用性,分别在3个不同实验室进行了方法验证实验,实验结果见表5。由表5可见,92%噁草酸原药、10%噁草酸乳油样品和标样重复性相对标准偏差分别为0.209 6%、1.559 5%,再现性相对标准偏差RSDR分别0.454 0%、2.034 4%,均小于相应的Horwitz公式理论计算值,表明不同单位间的检测结果符合性良好,本文建立的噁草酸液相色谱分析方法可满足日常检测工作需要。

表5 不同实验室方法验证试验结果
3 结 论
利用超高效液相色谱-二极管阵列检测器,甲醇溶解,检测波长235 nm,甲醇-水作为流动相,等度洗脱,建立了在同一条件下噁草酸原药及乳油的测定方法。在选择提取溶剂时,既考虑溶剂本身的性质,又要结合农药的物理化学性质及样品的状况,噁草酸的溶解性,本实验选择甲醇作为提取溶剂溶解样品。
采用液相色谱配置的二极管阵列监测器进行紫外区全波长扫描,得出噁草酸的最大吸收波长,所选的波长要避开其他物质的干扰。本实验选用235 nm作为测量波长,干扰小,灵敏度较高,进行了色谱峰特异性检验(纯度检验),大多数情况下,纯度检验可辅助进行方法开发。本研究建立的噁草酸的测定方法,两种制剂中噁草酸的回收率、精密度、RSD值均满足要求,具有操作简单、节省时间、灵敏度高、准确性及重现性好等优点,适用于噁草酸原药及乳油中有效成分的分析测定。
猜你喜欢
理化检验-化学分册(2022年11期)2022-11-27
农药科学与管理(2022年6期)2022-08-03
云南农业科技(2022年1期)2022-01-27
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
今日农药(2017年7期)2017-08-09
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
今日农药(2014年12期)2015-03-30
食品工业科技(2014年13期)2014-03-11
城市建设理论研究(2012年4期)2012-03-23
