
色素失禁症患儿33例的眼底表现及临床分析
2022-03-30 11:50谢琳辉罗瑜琳
国际眼科杂志 2022年4期
乔 静,谢琳辉,罗瑜琳
0引言
色素失禁症(incontinentia pigmenti,IP)又称Bloch-Sulzberger综合征,是一种罕见的以皮肤损害为主并可伴有眼、中枢神经系统、骨骼、牙齿等多系统异常的X染色体连锁显性遗传性疾病[1-2]。IP所导致的神经系统及眼部病变可威胁患儿的生命安全并严重影响患儿的生活质量,但由于该病的发病率仅为0.2/100000~1/50000,人们对该疾病缺乏足够认识,往往仅专注于患儿的皮肤及神经系统表现,忽略了眼部病变的检查及诊治,导致部分患儿错过最佳治疗时机而致盲[3]。因此,为了提高对IP患儿眼底病变的认识,了解病变的临床特点及发展趋势,为临床工作提供一定的依据,本研究回顾性分析在我院眼科就诊的33例IP患儿的眼底情况,结果报道如下。
1对象和方法
1.1对象收集2014-01/2018-12在我院确诊并于眼科就诊的IP患儿33例。IP诊断标准参照1993年Landy和Donnai提出的IP诊断标准[4],根据有无阳性家族史将患儿分为两类:(1)有阳性家族史者临床诊断标准:1)典型皮肤损害表现,包括色素沉着,原色素沉着部位皮肤相关瘢痕,皮肤条状无毛发,秃发;2)牙齿异常;3)视网膜疾病;4)多次妊娠男胎流产。符合以上1条临床诊断指标即可确诊。(2)无阳性家族史者主要诊断标准:1)新生儿期典型的皮肤红斑、水疱,水疱内含嗜酸性粒细胞;2)典型躯干部线状色素沉着;3)皮肤线状萎缩或秃发。次要诊断标准:1)牙齿异常;2)秃发;3)指甲异常;4)视网膜病变。至少需要1条主要诊断标准及1条次要诊断标准方可确诊。我院根据以上诊断标准,并结合基因检测结果或皮肤病理活检结果进行诊断,所有确诊患儿至少满足基因检测或皮肤病理活检中1项支持IP诊断。本研究符合湖南省儿童医院临床研究伦理学标准并获得伦理委员会批准,与患儿监护人均签订知情同意书。
1.2方法仔细询问并记录患儿母亲孕产史、患儿家族史、出生史及个人史,检查并记录患儿全身发育情况,如皮肤、神经系统、毛发、牙齿等。采用Retcam3系统对患儿进行眼底检查并根据眼底结果建议其定期随访,眼底病变严重需要干预者根据眼底具体情况给予视网膜激光光凝治疗、玻璃体腔注药及玻璃体切除术治疗。
1.2.1眼底病变分期标准目前对于IP相关性视网膜病变尚缺乏公认的分级标准,本研究参照赵培泉教授团队推荐的分期标准[5],将眼底病变分为5期:1期仅有视网膜色素上皮改变;2期为出现视网膜血管异常,但不伴有新生血管;3期为伴有新生血管的视网膜病变或其他继发于视网膜血管的病变,如视网膜渗出、视网膜前膜、视网膜增殖或玻璃体出血;4期为出现视网膜脱离(4a期:视网膜部分脱离;4b期:视网膜全部脱离);5期为终末期,伴有严重的眼部并发症,如眼球痨及继发性青光眼。
1.2.2随访原则参照Holmström等[6]和O’Doherty等[7]关于IP患儿眼底随访原则建议:对于确诊的IP患儿尽早进行眼底筛查,若眼底完全正常者每月检查1次持续至3月龄,然后每3mo检查1次持续1a,接着每6mo检查1次持续至3岁,3岁后眼底若无异常则可进行常规屈光眼保健随访;若首次眼底检查发现异常,建议尽早完善眼底血管荧光造影(fundus fluorescence angiography,FFA)检查明确视网膜缺血范围,并根据视网膜病变分级及干预治疗方法,在主治医师的指导建议下定期随访。由于监护人拒绝、担心麻醉风险等,本研究纳入患儿中仅有2例手术患儿行FFA检查。本研究对所有纳入患儿进行定期门诊复查及电话随诊,随访时间为1a。
2结果
2.1患儿基本资料及全身情况本研究纳入IP患儿33例,其中女31例,男2例,均为足月儿,母亲有流产史者9例(27%),患儿有IP家族史者2例(6%),均在出生后1wk内发病。根据患儿病史及临床表现,参考基因检测结果或皮肤病理活检结果进行确诊,确诊后定期随访皮肤、神经系统、毛发、牙齿,重点监测眼部表现,随访过程中1例患儿因皮肤感染致败血症死亡,1例患儿因重症神经系统疾病死亡(同时存在IP相关性视网膜病变)。纳入患儿全身情况见表1,均伴有不同程度皮肤损害(图1),其次为眼部异常(其中玻璃体视网膜病变14例、角膜混浊者1例、白内障1例、斜视2例)。
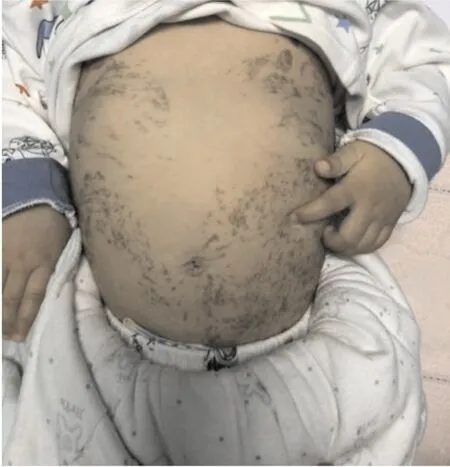
图1 IP患儿的皮肤改变 躯干部可见线样色素沉着。

表1 纳入患儿全身情况及辅助检查异常表现
2.2患儿初诊及随访时眼部表现纳入患儿33例初诊年龄5d~23月龄,平均年龄3.38±5.02月龄,确诊眼部异常者14例,其中男1例,女13例;单眼受累者5例,双眼受累者9例;神经系统异常表现者6例,毛发异常者7例,牙齿异常者4例。眼部异常表现包括角膜混浊1例1眼;白内障1例1眼;外斜视2例2眼;视网膜病变14例23眼(图2),其中1期病变2眼(表现为视网膜色素缺失或沉着),2期病变12眼(表现为视网膜血管迂曲、动静脉吻合),3期病变6眼(表现为视网膜出血、渗出,视网膜前膜、增殖性病变和玻璃体出血),4期病变3眼(表现为视网膜部分脱离或全部脱离)。3例患儿有眼部手术指征,1例患儿家长拒绝手术在随访过程中因神经系统症状加重而死亡,1例患儿行右眼玻璃体切除+左眼FFA检查联合视网膜激光光凝术,1例患儿行右眼玻璃体切除联合晶状体切除+左眼FFA检查联合视网膜激光光凝术。其余11例患儿基本按要求定期随访,末次随访时1例患儿眼底检查示IP相关性1期视网膜病变,其余患儿眼底病变消退。

图2 IP相关性视网膜病变眼底照相 A:1期,视网膜散在色素沉着(黄箭头);B:2期,视网膜异常血管吻合(黄箭头);C:3期,视网膜新生血管、视网膜出血(黄箭头);D:4b期,视网膜全脱离。
3讨论
由于皮肤损害是IP患儿的主要表现,往往在新生儿出生1wk内出现病变[8],易于被监护人及医护人员发现。但有文献报道约35%~77%的IP患儿伴有眼部异常,主要表现为眼底病变,如视网膜色素异常、血管异常甚至玻璃体出血、视网膜脱离[5,9-10]。此外IP患儿出现角膜异常、斜视、白内障、黄斑及视神经受累在临床上也均有报道[11-13]。不同研究报道的IP患儿眼部异常发生率各有不同,2013年我国学者报道IP患儿眼部异常的发生率仅24.2%[14],低于国外报道的水平,而2019年赵培泉教授团队报道IP患儿眼部异常的发生率高达77%[5]。本研究发现,在随访观察的33例确诊IP患儿中,眼部受累比例高达42%,且均存在眼底病变(14例23眼)。其次本研究纳入的IP患儿双眼不对称受累者居多,也可表现为单眼受累。不同研究中IP患儿眼部异常阳性率的差别可能与回顾性偏倚和转诊偏倚有关,同时,我们认为这与近年来儿科、眼科医师以及患儿家长对该病认识加深有关,也与目前小儿眼科筛查仪器及水平的进步有关。因此,对确诊的IP患儿进行规范的筛查与随访至关重要。
近年来随着对IP患儿眼底表现的临床资料进行不断积累和总结分析,对该病的发病机制、眼底特征及病程进展有了一定的认识。临床中发现多数IP患儿的眼底病变可自行停止在疾病的任一阶段,遗留视网膜色素改变、血管病变等,少数患儿的眼底病变会不断进展加重,甚至出现视网膜脱离、眼球萎缩[2,15-17]。根据赵培泉教授团队推荐的IP相关视网膜病变分期标准,本研究中初诊时患儿视网膜病变分级以轻度(1期和2期)为主(14/23,61%),随访过程中多数病变较稳定,仅2例进展型病例随访期间眼底病变发生进展,其FFA提示视网膜新生血管形成,发生玻璃体出血,均伴有严重的癫痫病。IP为多系统疾病,据报道30%患儿存在神经系统病变[3],多表现为惊厥、运动麻痹、抽搐及运动、语言、智力发育落后等,也是IP患儿的首位致死原因[18-19]。本研究中,14例IP相关性视网膜病变患儿有神经系统表现者6例,同时我们发现IP相关性视网膜病变患儿中神经系统受累者居多,这与Landy等[4]研究结果相一致,提示对于存在神经系统受累的IP患儿应格外重视眼底检查。
由于缺乏对新生儿及婴儿眼底血管荧光造影检查的设备以及手持式光学相干断层扫描仪器,本研究仅对患儿进行眼底彩色照相检查,未进一步评估患儿视网膜血管及细微结构的改变。由于IP相对罕见,本研究具有纳入病例数较少、临床治疗方案欠统一等局限性,未来我们将继续随访这些患儿,评估IP及相关眼底病变是否会影响远期视网膜发育及屈光状态,为临床上认识及诊断处理该类疾病提供一定的参考依据。
猜你喜欢
娃娃乐园·综合智能(2022年9期)2022-08-16
中国现代医生(2022年6期)2022-04-23
现代仪器与医疗(2021年4期)2021-11-05
中医眼耳鼻喉杂志(2021年1期)2021-07-22
科学大众(2021年9期)2021-07-16
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
疯狂英语·读写版(2019年6期)2019-09-10
家庭医药(2016年4期)2016-05-04
中国化妆品·下半月(2006年7期)2006-07-27
