
Tc1/Mariner转座子超家族在褶皱臂尾轮虫中的进化和表达分析❋
2022-03-26 02:10范正杰王玉珏张全启
中国海洋大学学报(自然科学版) 2022年4期
范正杰,王玉珏,张全启,2,3❋❋
(1.中国海洋大学海洋生物遗传学与育种教育部重点实验室,山东 青岛 266003;2.青岛海洋科学与技术试点国家实验室 海洋渔业科学与食物产出过程功能实验室,山东 青岛 266237;3.中国海洋大学三亚海洋研究院 海南省热带水产种质重点实验室,海南 三亚 572000)
转座子(Transposons),又被称为转座元件(Transposable elements, TEs),是一段能通过非同源重组在宿主基因组内或基因组间进行移动的散在分布的重复序列[1]。真核生物的转座子按转座机制可分为两大类:Ⅰ类元件即逆转录元件,是通过逆转录酶使用“复制-粘贴”的机制进行转座的RNA转座子;Ⅱ类元件DNA转座子,则是使用基于DNA的“剪切-粘贴”转座模式直接将转录子从原始位点整合至目标位点的转座子,其中最大的亚纲是两端有末端反向重复序列(Terminal inverted repeat,TIR)结构的TIR转座子,包含着分布广泛且种类众多的超家族,例如Tc1/Mariner超家族。Tc1/Mariner转座子超家族可能是自然界中分布最广的转座子超家族,在轮虫、真菌、植物、鱼类和哺乳动物等多种生物中都有其存在,然而其绝大多数是失活突变[2-4],且它因Tc1和Mariner合称而得名[5]。其中Tc1转座子于秀丽隐杆线虫(Caenorhabditiselegans)基因组研究时被发现[6],Mariner是最小的自主DNA转座子之一,以其水平转移的倾向而闻名,目前已经发现其在动物、植物、真菌中均广泛分布,即使在蛭形轮虫这类具有高转座抑制的物种中,Mariner转座子也通过其强大的不依赖宿主的特性而大量的存在与繁殖[4]。
Tc1/Mariner转座子的长度在1~5 kb之间,编码282~345个氨基酸的转座酶,其两侧有2个TIR,且TIR长度在17~1 100 bp之间[3,7]。来自不同Tc1/Mariner转座子家族的转座酶蛋白序列并不十分相似,但都具有两个特征结构域:一个包含螺旋-转角-螺旋(Helix-Turn-Helix, HTH)氨基末端,用于识别和结合TIR;另一个包含DDD/E羧基末端催化基序结构域,其第一和第二个天冬氨酸(D)残基之间至少有92个氨基酸,而第二个天冬氨酸(D)残基和第三个天冬氨酸(D)残基或谷氨酸残基(E)之间的距离是变量。目前Tc1/Mariner超家族大致可根据此数目分为7个主要亚家族,分别为DD34E(Tc1)、DD34D(mariner)、DDxD(pogo)、DD37D(maT)、DD37E、DD39D和DD41D(rosa)。
Tc1/Mariner转座子不依赖宿主的特征来执行转座过程,因此其存在不局限于一个特定的宿主。事实上,已经发现了许多Tc1/Mariner在不同宿主之间水平转移的情况,例如在褐带卷蛾中发现的一个Mariner转座子通过水平转移方式转移到它的寄生体后,也在其它寄主和寄生体间发现了Mariner转座水平转移的现象,表明寄生—寄主关系可能在转座子水平转移中占重要地位[8]。该现象存在于海洋甲壳类动物之间[9]、不同目昆虫之间[10-11],甚至存在于不同门的生物之间,如人类和寄生线虫之间的转移[12]。然而,目前还不清楚转座子是如何侵入新的基因组的。参与这种水平转移的潜在载体是外部寄生者,如螨虫(可能是果蝇中P元件水平转移的载体[13])?还是内部寄生者,如病毒[14]?目前针对Tc1/Mariner的水平转移在昆虫中报道较多[15]。
转座子在基因组中的分布并非是随机的,部分转座子与一些功能元件有密切的联系[16-17],转座子不仅影响了基因组的结构,还参与了基因表达调控的过程,例如在人类和小鼠中发现Alu插入的数量与基因差异表达存在相关性[18]。在大鼠和小鼠中发现,长末端重复序列(Long terminal repeated,LTR)和长散在重复序列(Long interspersed nuclear elements,LINE)与基因表达正相关[19]。此外,转座子插入到基因或基因侧翼区域也可能带来基因功能突变[20]。
褶皱臂尾轮虫(Brachionusplicatilis)在分类学上隶属于轮虫动物门(Rotifera),单巢纲(Monogononta)、游泳目(Ploimida)、臂尾轮虫科(Brachionidae)、臂尾轮虫属(Brachionus),是目前唯一能在海水养殖中实现规模化养殖的轮虫,具有抗逆性强、营养丰富、繁殖快和游动慢等特点,因此该物种成为海水鱼虾蟹类幼体重要的开口饵料[21]。同时褶皱臂尾轮虫具有典型的孤雌生殖世代和有性生殖世代交替的兼性繁殖生活史,会在外界环境变化,如温度、营养、种群密度和pH溶解氧剧变时进入混交世代。先前的转座子对于轮虫门基因组影响的研究大多关注含量及结构[22],而本研究基于褶皱臂尾轮虫基因组数据,针对褶皱臂尾轮虫基因附近的转座子家族进行鉴定和富集,并针对其Tc1/Mariner超家族进行了进化和表达分析,以从功能的视角去探索Tc1/Mariner超家族在褶皱臂尾轮虫基因组中扮演的角色。
1 材料与方法
1.1 褶皱臂尾轮虫基因组重复序列注释
褶皱臂尾轮虫基因组数据(Bioproject:PRJNA 719948)和基因注释信息由本实验室分析获得。重复序列注释方法分为同源序列比对和从头预测两类。同源序列比对方法基于RepBase[23],使用RepeatMasker[24]和Repeatproteinmask[25]软件识别与已知重复序列相似的序列。从头预测使用LTR_FINDER[26],Piler[27],RepeatScout[28],RepeatModeler[29]等软件:首先建立从头测序重复序列库;再通过Repeatmasker 软件预测。此外,使用TRF[30](http://tandem.bu.edu/trf/trf.html)寻找基因组中串联重复序列(Tandem repeat)。重复序列的注释均使用软件默认参数。将RepeatModeler、RepeatScout、Piler、LTR_finder 软件预测出来的结果结合RepBase 库采用Uclust[31]的软件(遵从80-80-80 原则)进行整合获得最终注释结果。
1.2 转座子及邻近基因表达分析
褶皱臂尾轮虫转录组数据(Bioproject: PRJNA 720972)由本实验室测序分析获得,取自8个发育阶段:非混交胚胎、非混交雌性、雄性、雄性胚胎、携带雄性胚胎的混交雌性、携带休眠卵的混交雌性、新产休眠卵、休眠三个月后的休眠卵(见图1)。使用Genomic-Ranges软件[32]及rtracklayer软件[33]对转座子及基因结构进行了统计,并且以基因两侧的5 000 bp的长度分别作为基因的上游和下游部分。转录组数据进行reads过滤,并使用STAR软件[34]进行比对得到SAM文件,再使用Samtools软件[35]转换为按名字排序的BAM文件并用于后续的分析。将基因注释、转座子注释文件和各个时期转录组进行比对结果,使用TEtranscripts软件[36]进行counts统计,并使用R语言软件中的DEseq2软件[37]进行标准化及处理,使用dplyr、factoextra[38]、FactoMineR软件[39]进行平行组及各个组之间的主成分分析以确保质量,最后用TBtools软件[40]来得到各个发育时期的所有转座子及基因的FPKM表达量。规定只有在各个发育时期FPKM≥3时认为该转座子有表达,进行筛选后将有表达的转座子使用R语言软件中的clusterProfiler软件[41]进行富集分析以获得转座子家族,对富集到的转座子家族与临近基因的各世代表达进行Person相关性分析,以绝对值0.5作为相关的系数标准,使用R语言软件中的ggplot2[38]进行绘图。
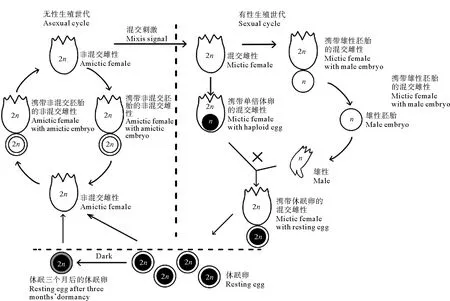
图1 褶皱臂尾轮虫生活史模式图
1.3 Tc1/Mariner转座子超家族的表达和进化分析
利用shell脚本提取褶皱臂尾轮虫基因组中的Tc1/Mariner转座子超家族注释信息,并分别统计其家族种类、拷贝数和表达量,最后对临近基因的功能进行分析。
为了获得基于Tc1/Mariner转座子完整转座酶,从NCBI及Repbase中下载了544条Tc1/Mariner转座酶序列,以褶皱臂尾轮虫基因组为目标库,使用tBlastN寻找转座酶序列(相似度>30%、长度>80、e值<10-5),对每个拷贝的两侧分别延长1 000 bp,使用Seqkit[42]的subseq功能基于bed格式提取完整fasta序列,使用TBtools中的ORF_Prediction获取序列的ORF序列,并保留每个序列预测出的长度大于300 bp的氨基酸序列,在MuscleX软件中使用对齐(即使用默认设置),手动进行筛选,保留有完整的DDE/D结构域的转座酶序列,并基于DDE/D结构域确认其家族,在PHYRE2网站(http://www.sbg.bio.ic.ac.uk/phyre2)上预测各家族转座酶的空间结构。
使用已知的具有完整DDE结构域的转座酶家族(来自于果蝇、蛭形轮虫、家蚕等)作为参考,使用muscle软件[43]进行对齐,用ProtTest软件[44]选择最优氨基酸代替模型,基于该模型使用RAxML软件[45]构建系统进化树,bootstrap设为1 000。对于获得的系统树,使用EvolView软件[46]进行美化。
利用Tc1/Mariner各个亚家族的注释信息,使用TEtranscripts软件处理结果来得到各个时期的表达量。用GenomicRanges软件及rtracklayer软件寻找各家族附近基因,并基于基因功能注释得到Tc1/Mariner各亚家族附近基因的功能富集结果。
2 结果
2.1 褶皱臂尾轮虫基因附近转座子家族富集分析
主成分分析结果显示(见图2),8个组24个转录组聚类情况良好,可用于后续分析。褶皱臂尾轮虫基因组中共鉴定到419 244个转座子(不含简单重复序列),从中筛选得到了92 225个转座子在褶皱臂尾轮虫的生命周期中的不同阶段发生了表达。共富集到10个转座子家族中,其中Ⅰ转座子家族有3个:ERV4、Pao和tRNA-Deu;Ⅱ转座子家族有7个:包括隶属于Tc1/Mariner超家族的Mariner、Fot1和Tigger3个亚家族,以及不属于Tc1/Mariner超家族的hAT1、hAT、Academ和Sola。将这10个转座子家族的转座子拷贝表达量与邻近基因的表达量进行相关性分析,发现多数转座子的表达模式与附近基因的表达模式呈正相关,负相关者较少,其中呈相关性的Mariner家族的转座子以分布在基因下游为主(见图3a),而Fot1家族的转座子以mRNA分布居多(见图3b),Tigger家族的转座子则分布较为均匀(见图3c)。
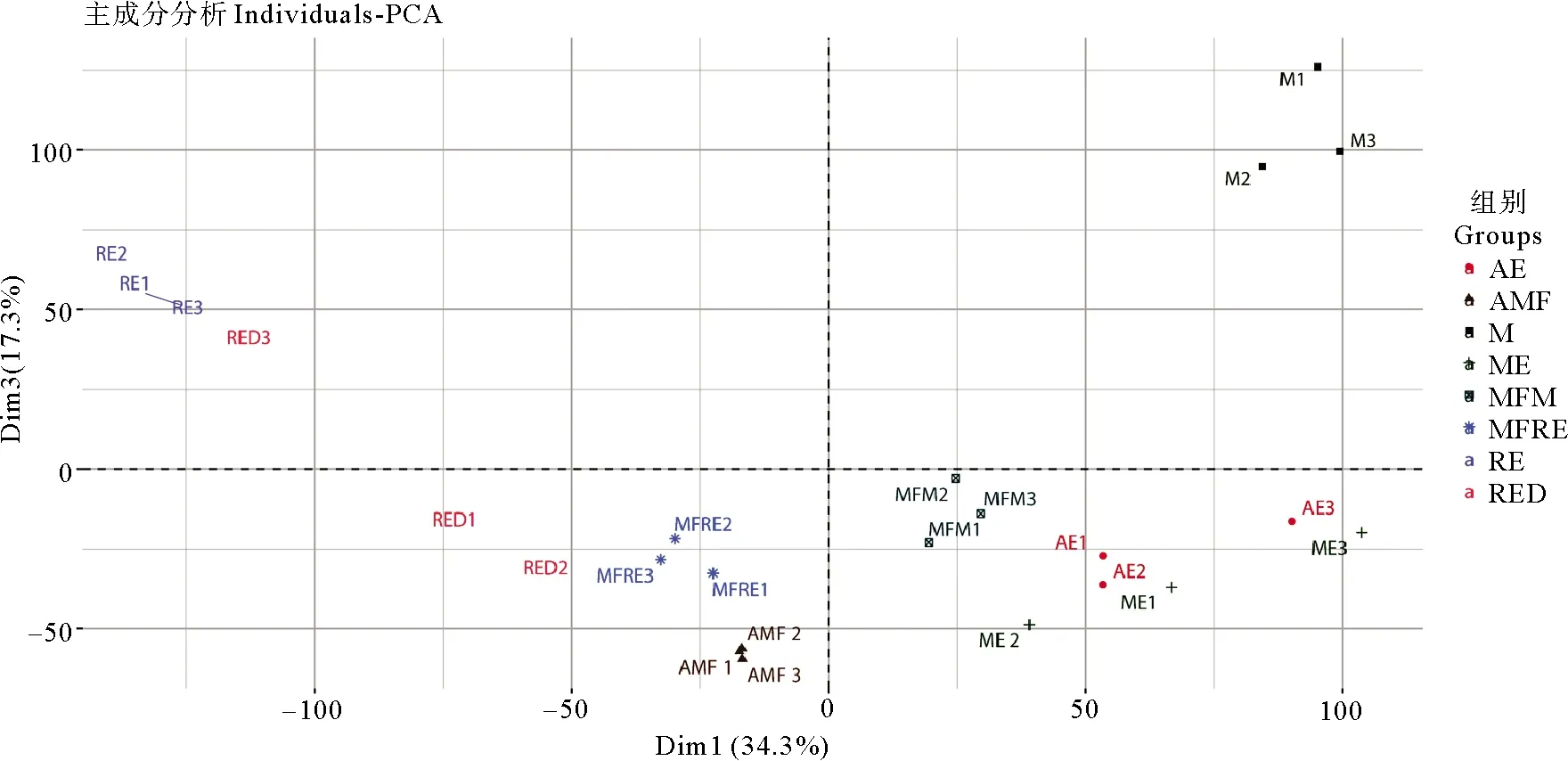
(AE: 非混交胚胎Amictic embryos;AMF: 非混交雌性Amictic females;M: 雄性Males;ME: 雄性胚胎Male embryos;M: 携带雄性胚胎的混交雌性Mictic females with male embryos;MFRE: 携带休眠卵的混交雌性Mictic females with resting eggs;RE: 新产的休眠卵Resting eggs;RED: 休眠三个月后的休眠卵Resting eggs after three months’ dormancy)

((a)Mariner家族Mariner family;(b)Fot1家族Fot1 family;(c)Tigger家族Tigger family。第一列为Pearson相关性系数,第二列-log10(p值),第三列转座子位置,第四列为转座子FPKM表达量,第五列为转座子附近基因FPKM表达量。1st column: correlation; 2nd column:-log10(p-value);3rd column: TE position; 4th column: TE FPKM expression level; 5th column: nearby genes’ FPKM expression level.AE: 非混交胚胎Amictic embryos;AMF: 非混交雌性Amictic females;M: 雄性Males;ME: 雄性胚胎Male embryos;M: 携带雄性胚胎的混交雌性Mictic females with male embryos;MFRE: 携带休眠卵的混交雌性Mictic females with resting eggs;RE: 新产的休眠卵Resting eggs;RED: 休眠三个月后的休眠卵Resting eggs after three months’ dormancy)
2.2 Tc1/Mariner的鉴定和转座酶结构及系统发育分析
褶皱臂尾轮虫中共发现了23 378个Tc1/Mariner转座子拷贝,并分为7个亚家族,其中拷贝数目较多的3个家族为Tc1、Pogo和Mariner家族(见表1)。
统计了7个亚家族在基因内部、基因上下游及基因间区的分布情况(见表1)。所有亚家族表现出了相似的分布特征,即均在基因间区分布最多。除了Trigger亚家族(48.19%)和Tc1亚家族(47.73%),其余5个亚家族在基因间区的转座子分布占比都超过了50%。Sagan亚家族在基因间区的分布占比最高(65.69%)。在基因内部分布的情况中可以发现,相较于其他6个亚家族,Sagan亚家族在基因内部的分布最少(仅占2.93%)。
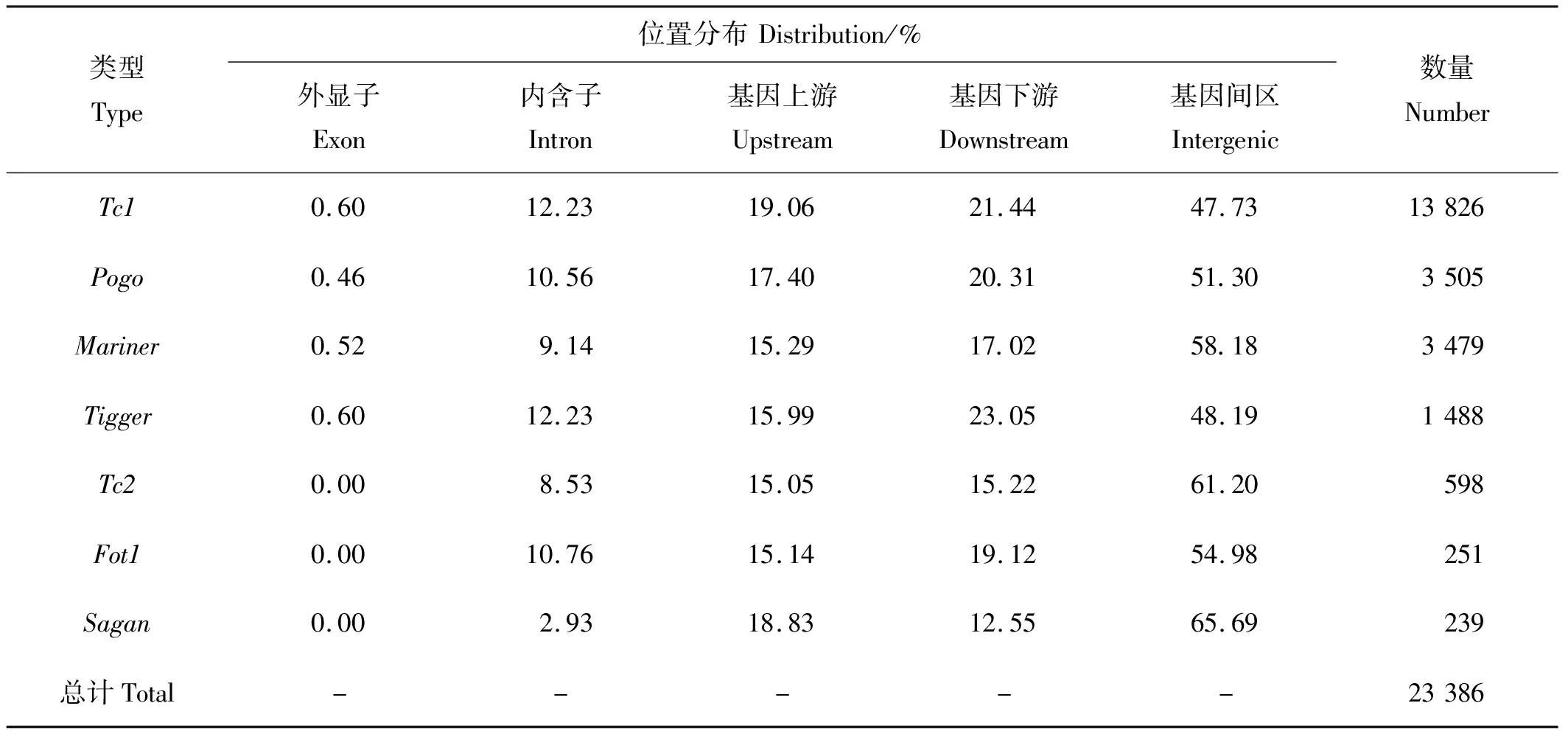
表1 Tc1/Mariner转座子7个亚家族的位置分布及拷贝数
基于同源比对的方法,本文作者在褶皱臂尾轮虫基因组中共发现了29条完整的Tc1/Mariner转座子序列。为了确定褶皱臂尾轮虫中Tc1/Mariner转座子的系统发育关系,基于转座酶序列使用Repbase和NCBI中下载的近源物种中的Tc1/Mariner转座子基因序列进行了系统发育分析。结果发现褶皱臂尾轮虫中的Tc1/Mariner转座子可以分为3个不同的亚家族即Tc1、Tc2和Pogo,其中大多数为Pogo转座子(见图4)。系统发育分析结果表明,褶皱臂尾轮虫Tc1亚家族与家蚕的Tc1亚家族关系较近,而与果蝇中的Tc1家族关系较远;Tc2亚家族与秀丽隐杆线虫的Tc2聚为姐妹群;Pogo亚家族与真涡虫Pogo亚家族关系较近。总体来看,这与褶臂尾轮虫Tc1/Mariner转座子系统发育关系与物种之间的亲缘关系基本一致。

(不同的背景颜色代表不同的Tc1/Mariner转座子亚家族,红色分类名代表褶皱臂尾轮虫中鉴定到的序列。Clade colors denote different subfamilies of Tc1/Mariner transposons; Taxon names in red indicate sequences from B. plicatilis.)
然后对29条完整转座酶进行了结构预测和序列比对。Tc1/Mariner转座酶的N端为DNA结合区域,C端为催化区域,空间结构预测结果(见图5)表明褶皱臂尾轮虫的Tc1/Mariner转座酶均在Phyre2中高置信命中了c3hosA(转座酶)结构,其N端NA结合区域含有两段螺旋-转角-螺旋(HTH,helix-turn-helix)结构,而在转座酶的C端均具有DDE/D基序作为DNA剪切转座活性的催化区域。序列对比结果显示Tc1/Mariner超家族转座酶催化区域的第二个天冬氨酸(D)和第三个天冬氨酸(D)之间,或第二个天冬氨酸(D)和谷氨酸(E)之间有30~35个氨基酸,基于其数目不同可以分为不同的家族,分别是Tc1家族(DD34E,见图6a)、Tc2(DD35D,见图6b)家族和Pogo(DDxD,见图6c)家族。

图5 Tc1/Mariner转座酶的空间结构预测
2.3 Tc1/Mariner转座子超家族的表达模式及功能
Tc1/Mariner转座子7个亚家族在各个发育阶段均有表达,并且具有相似的表达模式,但表达量并非恒定一致(见图7),在孤雌生殖阶段的非混交胚胎和非混交雌性中表达量偏低,在有性生殖阶段中,表达量较高,其中转座子在雄性阶段表达量最高,在携带雄性胚胎的需精雌性和携带休眠卵的混交雌性中表达量较低,在休眠卵(包括新产和休眠三个月)中的表达量较高。

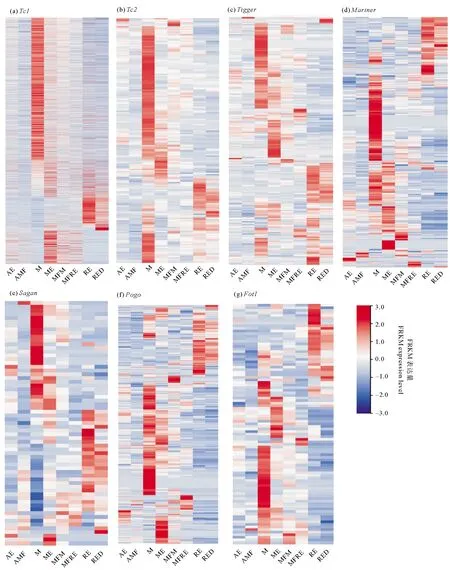
(AE: 非混交胚胎Amictic embryos;AMF: 非混交雌性Amictic females;M: 雄性Males;ME: 雄性胚胎Male embryos;MFM: 携带雄性胚胎的混交雌性Mictic females with male embryos;MFRE: 携带休眠卵的混交雌性Mictic females with resting eggs;RE: 新产的休眠卵Resting eggs;RED: 休眠三个月后的休眠卵Resting eggs after three months’ dormancy.)
为了了解各个亚家族临近基因的功能,本文作者对每个亚家族临近的基因进行GO和KEGG富集分析。
GO功能富集(见图8)发现了大量离子相关功能,包括离子结合和离子转运。其中离子结合功能包括锌离子结合和镁离子结合等,而离子转运涉包括阴离子转运等。同时也发现了细胞周期调控功能,包括细胞周期功能DNA修复功能和多细胞生物发育的调节功能。此外还发现应对刺激的功能,包括在应对外界压力和生物过程的调控的功能,以及损伤的反应。
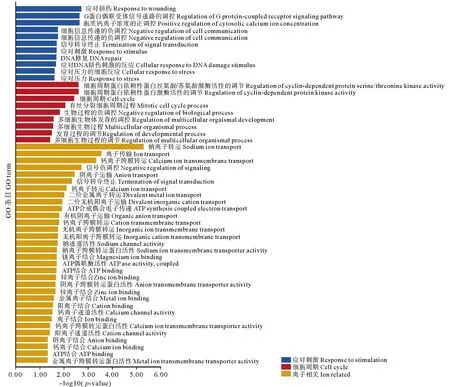
图8 筛选出Tc1/Mariner临近基因的GO功能富集
KEGG通路富集(见图9)中发现的细胞周期通路和环境信息处理的通路有大量的富集。其中大量富集于细胞周期的通路包括减数分裂和衰老通路。大量富集于环境信息处理的通路包括鞘脂信号通路、Notch、Jak-STAT、HIF1、TGF-beta及Wnt信号通路等。
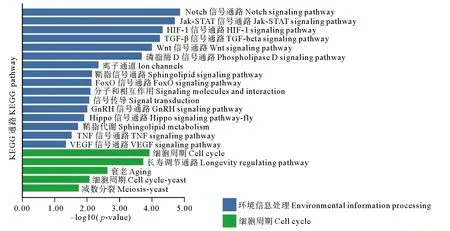
图9 筛选出的Tc1/Mariner临近基因的KEGG信号通路富集
总之,Tc1/Mariner转座子超家族附近基因中有大量涉及离子相关、应对刺激、环境信息处理及生物发育调节的功能和通路。
3 讨论
通过统计褶皱臂尾轮虫基因组中的转座子的分布,发现分布在基因内含子区域的转座子较多。相较于基因编码区域,内含子区域的转座子受到的选择压力更小,从而获得相较于其他区域更高的存活机会[47],同时内含子区域的转座子插入可能影响mRNA的可变剪切及转录产物的正确加工。但是目前还没有足够的证据证明褶皱臂尾轮虫中转座子的插入位置偏倚是随机插入后受到选择压力的选择结果,还是在转座子本身的偏好性带来的[48]。
转座子在基因组中的分布与基因表达的各个方面相关,包括表达水平、转录本多样性和基因调控因子的活性[49]。目前,关于转座子家族作为整体对基因表达的影响的研究还较少,有研究报道LINE1家族在小鼠原始生殖干细胞和着床前胚胎中RNA含量丰富并且其敲除会抑制原始生殖干细胞更新[50]。本研究通过使用生物信息学手段分析了转座子表达与轮虫各个生命阶段的相关关系,在发育的各个阶段都鉴定到了表达的转座子及富集的转座子家族。共富集到了10个转座子家族,其中DNA类型的转座子含优势,其中有Mariner、Fot1和Tigger3个亚家族均来自于Tc1/Mariner超家族。同时发现转座子的表达与临近基因的表达以正相关为主。
已有文献报道脊椎动物中Tc1/Mariner转座子具有多样性,其丰度各有差异[51]。Tc1/Mariner转座子超家族在哺乳动物和鸟类中表现出非常低的多样性和丰度[52-53],与哺乳类和鸟类中不同,早前的研究在硬骨鱼的基因组中观察到了Tc1/Mariner转座子的高多样性和高丰度[54]。例如转座子可以插入到基因组中的新的调控元件、外显子及内含子中介导基因融合和基因沉默[55]。而在Tc1/Mariner超家族中,许多转座子例如Tc1亚家族不仅可以参与基因表达调控,还可以促进新基因的产生,进而使物种适应新环境[56]。本研究发现Tc1/Mariner转座子在褶皱臂尾轮虫基因组中表现出多样性,共鉴定出7个Tc1/Mariner转座子亚家族。根据其催化区域第二个天冬氨酸(D)和第三个天冬氨酸(D)之间,或第二个天冬氨酸(D)和谷氨酸(E)之间的氨基酸数目将其鉴定为Tc1(DD34E)、Tc2(DD35D)、Mariner(DD34D)、Pogo(DDxD)、Sagan(DD30D)、Tigger(DD32D,DD36D)和Fot1(DD30D)。然而,在自然界中发现的绝大多数Tc1/Mariner转座子被认为是有缺陷的,因为它们在转座酶基因的编码区域内包含了移码、插入/缺失和过早终止密码子,因此很少有转座子(如Passport和Thm3)被认为是功能活性元件[57-58]。在哺乳动物和鸟类中,所有Tc1/Mariner转座子都是化石转座子且结构有缺陷[52-53,59]。在两栖动物中,Tc1/Mariner超家族的活动似乎也已经灭绝,尽管Tc1/Mariner超家族占青蛙基因组的很大比例(约5%),但大多数元件都是古老的,并且没有独立的家族被鉴定出来[60]。爬行动物中Tc1/Mariner多样性也很低,Tc1/Mariner超家族是蜥蜴DNA转座子中第二多的超家族,但只有一个独立的家族被鉴定出来[61]。在褶皱臂尾轮虫中,Tc1/Mariner转座子具有多样性,这可能是因为在淡水生态系统中,洪水和干旱等频繁的压力可以加快转座活动,从而有助于宿主产生新的变异以适应变化的环境[25, 62-63]。
Tc1/Mariner转座酶中最后两个残基之间的氨基酸数量在30~36之间。除去缺陷拷贝中的突变,大多数DD34E(Tc1)和DD34D(Mariner)元件包含典型的34个氨基酸间距(DD34E和DD34D)[64]。在本研究中,本文作者发现褶皱臂尾轮虫的DD34E(Tc1)中的大部分元件都表现出典型的DD34E 结构。在DDxD(Pogo)亚家族在不同物种中可以观察到距离的变化,例如真菌的Flipper、Pot2和Fot1中的DD35D[65-66]、果蝇的DD30D[25,67]、人类的DD32D(Tigger1)[68],而在硬骨鱼中,所有完整的转座子元件都显示出非常保守的DD35D 结构域[69]。本研究鉴定出的3个亚家族的29条完整转座酶序列也均具有良好的保守性,包括Tc1-DD34E、Tc2-DD35D和Pog-DDxD,这一结果表明,硬骨鱼类中Tc1/Mariner转座酶的活性可能有其特殊性。
对褶皱臂尾轮虫的Tc1/Mariner转座子的各个亚家族的各发育阶段的表达情况和临近基因功能进行了分析,发现各个亚家族的表达模式基本相似,均在雄性发育阶段表达量最高,在新产休眠卵和休眠三个月后的休眠卵的表达量较高,这2个时期均为轮虫发育状态发生变化的特殊时期,这期间转座子处于活跃状态,这为后续研究转座子在轮虫发育中的作用提供了重要的材料和线索。在对其临近基因功能分析中,各个亚家族的GO功能诸多涉及到多细胞生物发育的调节、离子通道、细胞周期、对刺激反应的调控、DNA修复等;KEGG分析也发现与环境信息处理、生物发育调节有关。这些都可能与环境适应有关,而在轮虫基因组中,DNA修复和离子转运属于轮虫适应环境比较独特的功能,涉及到神经调节、渗透压调节、细胞周期调节、无性世代有害变异修复和休眠卵滞育等一系列的生物学过程。显然,轮虫基因组对环境的适应是综合的调节,不局限于基因层面,还包含了转座子表达对基因表达的调控。因此,有关Tc1/Mariner转座子具体的生物学功能,需要进一步的实验去验证。
本研究通过对褶皱臂尾轮虫Tc1/Mariner转座子超家族在基因组中的分布、进化和表达进行系统分析,这为系统地认知Tc1/Mariner转座子并从功能角度理解其对褶皱臂尾轮虫基因组的作用提供了新的线索。
猜你喜欢
军事文摘(2022年16期)2022-08-24
今日农业(2022年4期)2022-06-01
中国典型病例大全(2022年7期)2022-04-22
学生导报·中职周刊(2019年7期)2019-06-11
电影(2019年3期)2019-04-04
阅读(低年级)(2018年11期)2018-05-14
少儿科学周刊·少年版(2017年3期)2017-06-29
Coco薇(2017年6期)2017-06-24
花样盛年(2016年8期)2016-09-07
Coco薇(2015年7期)2015-08-13
