
高效液相色谱法测定牛、羊可食性组织中莫昔克丁残留量
2022-03-24 13:28张玉洁黄耀凌孙红洋王鹤佳
中国兽药杂志 2022年2期
沈 昕,张玉洁,黄耀凌,李 丹,张 骊,孙红洋,王鹤佳
(中国兽医药品监察所,北京 10081)
莫昔克丁,也称莫西菌素,是一种安全、长效的杀虫药,在日本、欧洲等多个国家登记,并作为对环境友好的杀虫剂被美国环保局推荐使用[1]。研究表明,莫昔克丁能杀灭盘尾丝虫的微丝蚴,还可通过抑制成虫体内的胚胎发育和微丝蚴的释放发挥其作用,同时不同程度的降低各成长阶段盘尾丝虫的蠕动、成虫的繁殖能力以及免疫调节蛋白的分泌[2-3]。自20世纪80年代中期以来,莫昔克丁也被作为驱虫药广泛应用于畜牧业中[4]。相关文献表明,莫昔克丁对牛、羊、猪体内的消化道线虫、肺线虫和体外寄生虫都具有良好的驱杀作用,其与吡虫啉的混合制剂可用于治疗犬、猫的线虫病、螨虫病及钩端螺旋体炎,同时也能在禽类体内保持高效的驱虫活性[5-18]。2014年,吡虫啉莫昔克丁滴剂(猫用、狗用)被批准在国内上市,同年,由浙江海正药业股份有限公司研发的二类新药莫昔克丁及莫昔克丁浇泼溶液在国内获批上市。
莫昔克丁为第三代阿维菌素类药物,具有很高的亲脂性,在脂肪中分布极高,粪便为其主要排泄途径,且主要残留形式为莫昔克丁原药[14]。我国GB-31650-2019规定莫昔克丁在牛肌肉中的最大残留限量(maximum residue limit, MRL)为 20 μg/kg,在绵羊肌肉中的MRL为50 μg/kg,在牛、绵羊的脂肪、肝及肾中的MRL分别为500 μg/kg、100 μg/kg和50 μg/kg[19]。目前,我国尚无动物组织中莫昔克丁残留检测方法的国家标准,且同时针对多种动物组织中莫昔克丁的残留方法的报道也较少[21-23]。本研究拟建立一种适用于牛、羊组织样品中莫昔克丁残留检测的高效液相色谱方法,以满足当前的实验室检测需求,同时为动物食品安全检测提供有效技术手段。
1 材料与方法
1.1 材料与试剂 莫昔克丁标准品(纯度≥94%),德国Dr.Ehrenstorfer公司;、乙腈、甲醇均为色谱纯,三乙胺、三氟乙酸酐、冰醋酸,国药集团化学试剂有限公司;N-甲基咪唑,上海麦克林生化科技有限公司;水为一级水。
1.2 仪器与设备 高效液相色谱仪(1260,配荧光检测器),美国Agilent公司;分析天平(XS105DU),电子天平(AE260),Mettler Toledo公司;Biofuge stratos离心机,赛默飞世尔科技(中国)有限公司;涡旋混合器(MS3),振荡器(ks 501 digital),德国IKA公司;氮吹仪(FV64),得泰仪器公司;鼓风干燥箱(LC-213),上海爱斯佩克环境仪器有限公司;Milli-Q纯水仪,美国Millipore公司;C18固相萃取柱,美国Agilent公司。
1.3 方法
1.3.1 溶液配制
1.3.1.1 标准溶液配制及标准曲线绘制 按莫昔克丁标准品纯度折算后,精密称定10.0 mg于10 mL容量瓶中,用乙腈溶解并稀释至刻度,配制成浓度为1 mg/mL的标准储备液;准确吸取1.00 mL标准储备液于100 mL容量瓶中,用乙腈稀释至刻度,成为10 μg/mL的标准工作液,密封贮存于-20 ℃冰箱。
精密量取莫昔克丁标准工作液适量,分别用乙腈逐级稀释,制成浓度为4、10、100、500、1000和2000 ng/mL的标准工作液。以上6个浓度的标准工作液,分别准确吸取1.0 mL于10 mL试管中,50 ℃水浴下氮气吹干后,进行衍生化,加入甲醇,上机检测,绘制标准曲线。
1.3.1.2 衍生化试剂的配制 分别以N-甲基咪唑-乙腈(1+1,V/V)和三氟乙酸酐-乙腈(1+2,V/V)作为衍生化试剂A液、B液,并保证临用现配。
1.3.2 样品前处理
1.3.2.1 提取 称取均质后的牛、羊的肌肉、脂肪、肝脏、肾脏组织各(2±0.02) g置于50 mL离心管中,加入5 mL乙腈,涡旋混匀后中速振荡5 min,10000 r/min离心5 min,将上清液转移至另一50 mL离心管中。按照上述步骤再提取一次,将两次上清液合并,加入5 mL水,涡旋混匀后,加入三乙胺20 μL(羊肝脏加三乙胺50 μL),混匀备用。
1.3.2.2 净化 依次用5 mL乙腈和5 mL 30%乙腈水溶液活化C18固相萃取柱,将上述备用液全部过柱后,用5 mL 30%乙腈水溶液淋洗,用5 mL乙腈洗脱萃取柱。将洗脱液收集于10 mL具塞试管中,50 ℃水浴氮气吹干。
1.3.2.3 衍生化 依次向吹干后残渣中加入100 μL衍生化试剂A液和150 μL衍生化试剂B液,密闭涡动10 s后,加入50 μL冰醋酸和三乙胺各50 μL,漩涡混匀10 s,65 ℃下,避光密闭反应15 min。反应完成冷却后,加入650 μL甲醇,混匀后经0.45 μm微孔滤膜过滤,供高效液相色谱检测。
1.3.3 色谱条件 色谱柱:Phenomenonx luna C18250 mm × 4.6 mm(i.d), 粒径5 μm;柱温:40 ℃;进样体积:20 μL;流动相:乙腈+水(90+10,V/V);流速:1.8 mL/min;检测波长:激发波长365 nm,发射波长475 nm。
1.4 方法灵敏度的确定 分别在北京市内购置的不同品牌及厂家的牛及羊的肌肉、脂肪、肝脏、肾脏进行筛选,所得空白样品进行添加回收试验,按照上述样品前处理方法处理,进HPLC 分析,测得药物色谱保留处的基线噪音值,求其平均值。以S /N≥3作为该方法药物的检测限;以S /N≥10,且在该添加水平的回收率和变异系数均满足残留分析要求的最小浓度作为定量限(LOQ)。
1.5 准确度和精密度的测定 采用标准添加法,在空白牛及羊的肌肉、脂肪、肝脏、肾脏组织中分别添加定量限、1/2 MRL、MRL和2 MRL四个浓度的莫昔克丁标准工作液进行回收率试验,各浓度进行5个样品平行试验,同时重复3次空白试验,求每个样品的回收率和批内、批间相对标准偏差(RSD)。
2 结果与分析
2.1 线性范围与检测限和定量限 配制浓度分别为4、10、100、500、1000和2000 ng/mL的标准工作液,按照1.3.2项步骤处理,上机检测,得到莫昔克丁在6个浓度水平下的线性回归方程为y=0.3412x-3.2242,其相关系数大于等于0.9996,线性范围较好。依据信噪比S/N ≥3和S/N ≥10(按PtP计算),确定检出限(LOD)及定量限(LOQ)分别为2 μg/kg和5 μg/kg。标准对照液、空白牛、羊肝脏及添加标样中的高效液相色谱图见图1~图2。

图1 莫昔克丁标准溶液色谱图(10 ng/mL)
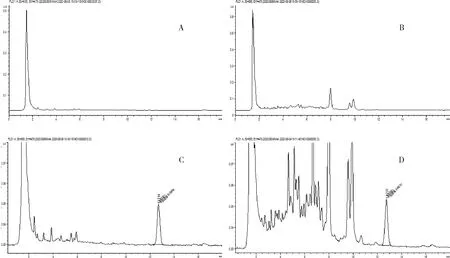
图2 空白牛肝脏试样色谱图A;空白羊肝脏试样色谱图B;牛肝脏添加莫昔克丁试样色谱图(5 μg/kg)C ;羊肝脏添加莫昔克丁试样色谱图(5 μg/kg)D
2.2 方法的准确度和精密度 本方法分别选用由北京本地市场和网上购买的样品进行空白样品筛选。分别在8种不同空白样品中添加其相对应的定量限、1/2 MRL、MRL和2 MRL水平浓度后进行测定,其中牛肌肉分别添加5、10、20、40 ng/g水平浓度,牛、羊肝脏分别添加5、50、100、200 ng/g水平浓度,牛肾脏、羊肌肉、羊肾脏分别添加5、25、50、100 ng/g水平浓度,牛脂肪、羊脂肪分别添加5、250、500、1000 ng/g水平浓度,每个水平浓度做5个平行样品,并重复3次实验,计算其批内、批间相对标准偏差,结果见表1~表8。

表1 牛肌肉中莫昔克丁添加回收率
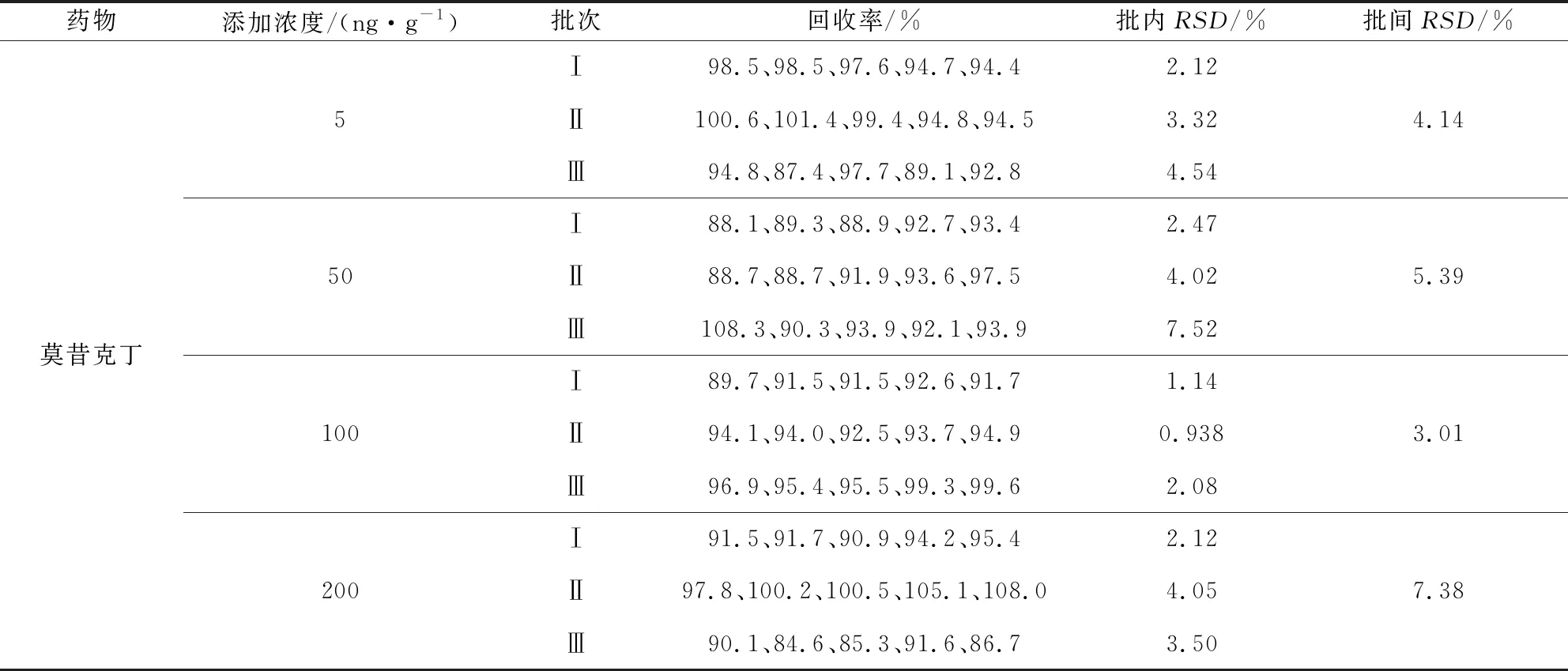
表2 牛肝脏中莫昔克丁添加的回收率
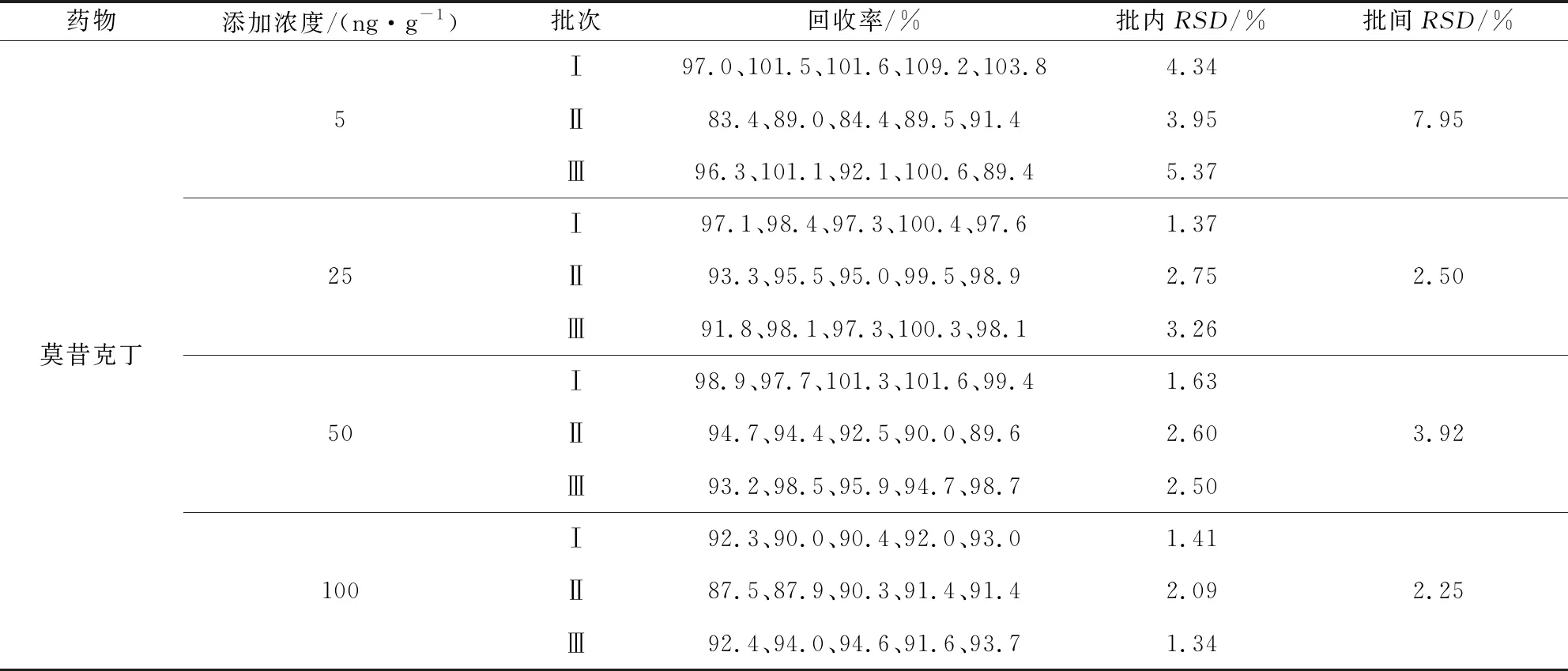
表3 牛肾脏中莫昔克丁添加的回收率
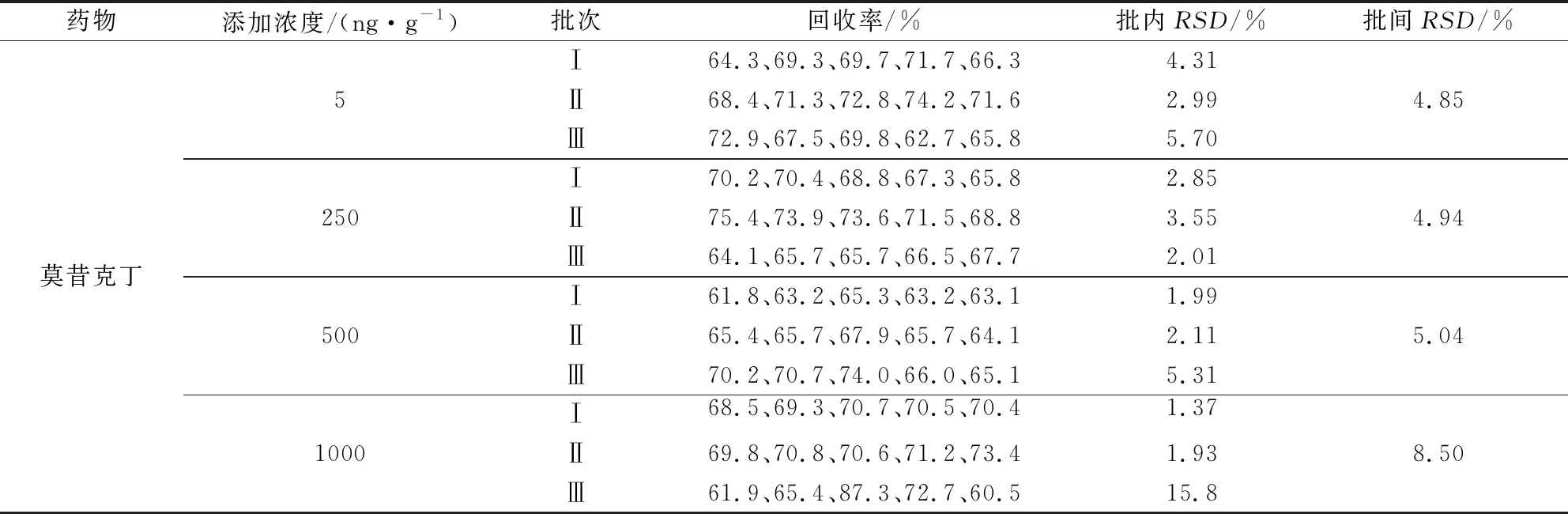
表4 牛脂肪中莫昔克丁添加的回收率
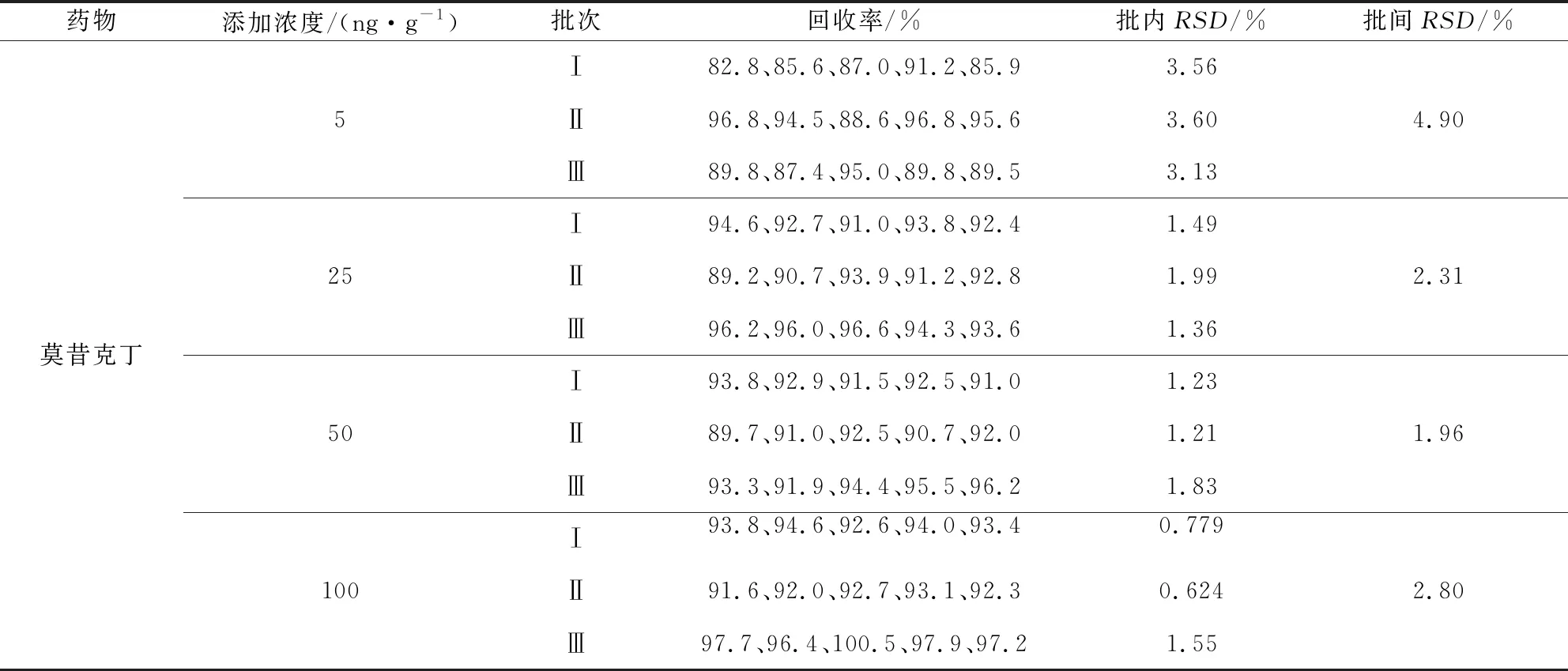
表5 羊肌肉中莫昔克丁添加的回收率

表6 羊肝脏中莫昔克丁添加的回收率
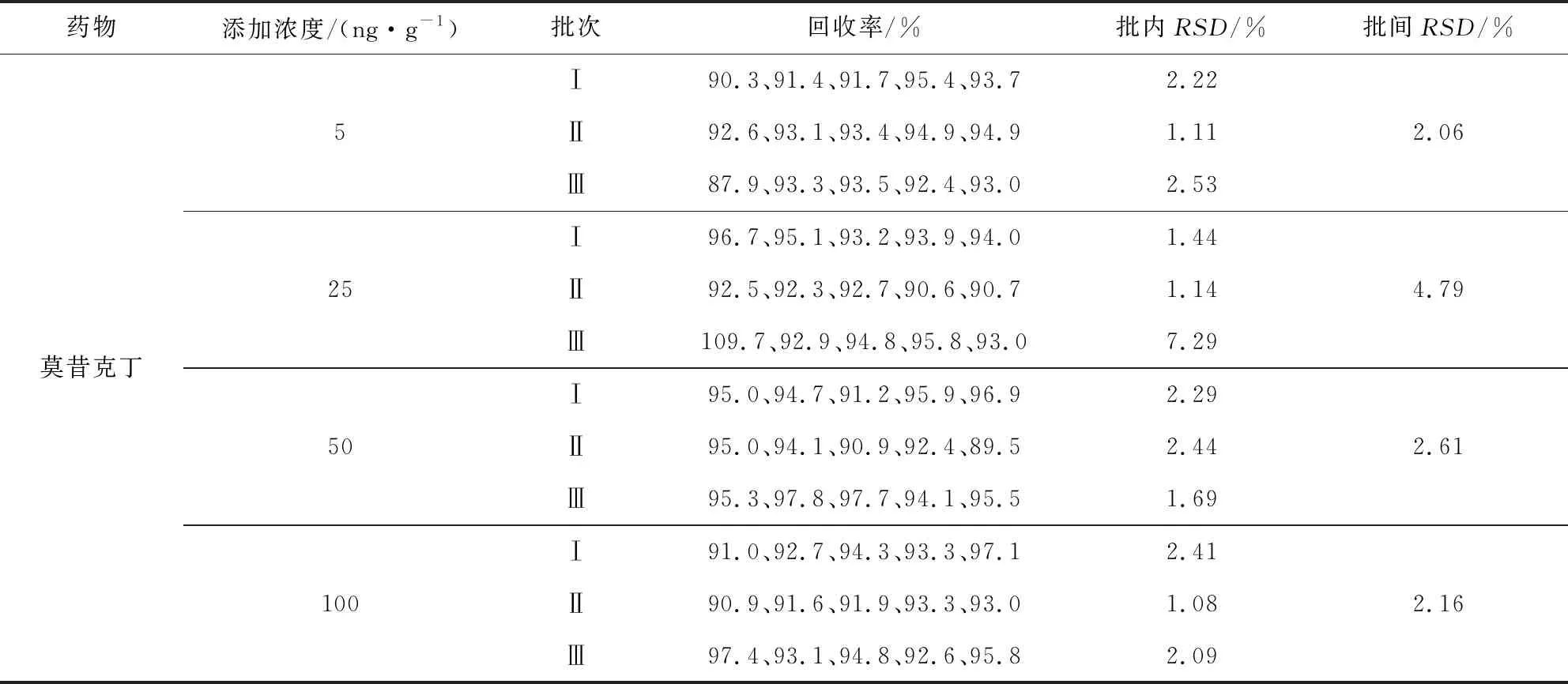
表7 羊肾脏中莫昔克丁添加的回收率

表8 羊脂肪中莫昔克丁添加的回收率
结果显示,莫昔克丁在牛肌肉的添加回收率为71.8%~103%,批内相对标准偏差≤10.6%,批间相对标准偏差≤7.55%;对羊肌肉、肾脏,牛肾脏的添加回收率均在82.8%~110%之间,批内相对标准偏差≤7.29%,批间相对标准偏差≤7.95%;对牛肝脏、羊肝脏的添加回收率均在74.7%~108%之间,批内相对标准偏差≤8.15%,批间相对标准偏差≤9.04%;对牛、羊脂肪的添加回收率均在60.5%~ 87.3%之间,批内相对标准偏差≤15.8%,批间相对标准偏差≤8.50%。
3 讨论与结论
3.1 提取条件的优化 莫昔克丁易溶于多种有机溶剂[16-18],本方法分别对乙酸乙酯、20%乙醇乙腈、乙腈的提取效果比较时发现,乙酸乙酯提取回收率较低,20%乙醇乙腈和乙腈提取回收率均能达到80%以上,但经比较分析,20%乙醇乙腈提取后,药物峰形变宽,因此,本方法选择乙腈作为提取溶剂,并通过高速离心去除蛋白质等大量杂质的干扰,有效提取其中的待测药物。
实验过程中发现,提取液中加入适量三乙胺可降低基质杂质的影响,但加入过多三乙胺会影响药物的回收率,因此,本方法经过向不同组织中分别添加不同量的三乙胺,最终确定合适添加量为20 μL,但羊肝脏中需添加50 μL,可达到较好效果(图3)。
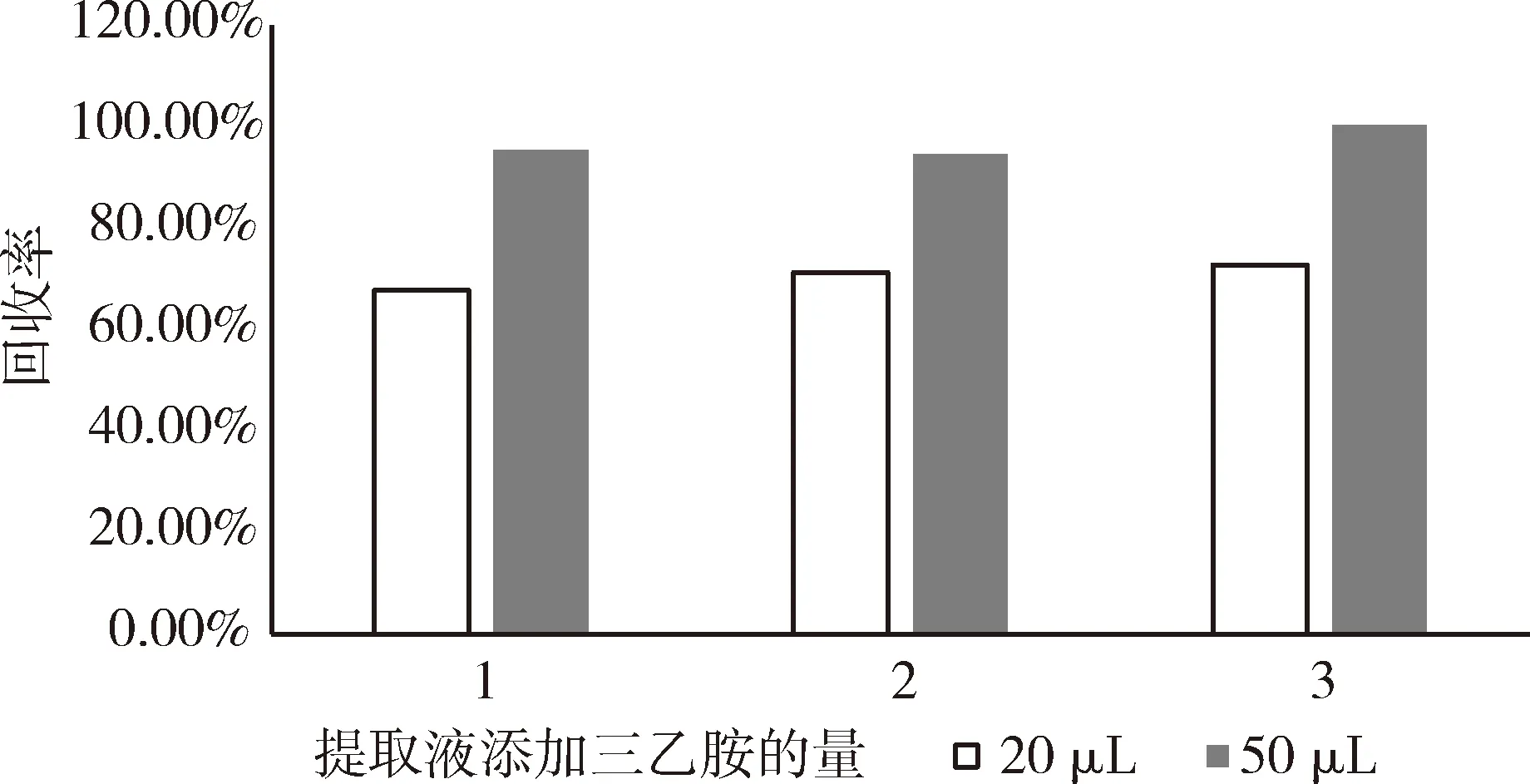
图3 提取溶剂对回收率的影响(羊肝脏)
3.2 净化条件的优化 由于莫昔克丁具有很高的亲脂性[17-18],同时,动物肌肉及内脏中含有丰富的脂肪,因此,本试验未选用正己烷等有机溶剂进行除脂。通过参考阿维菌素类药物净化方法的文献[20-26],试验分别选取HLB柱和C18柱进行净化比较。试验发现,虽然2种固相萃取柱回收率与稳定性均较好,但HLB柱过滤后的溶液杂质较多,C18柱的净化效果更好,且在去除蛋白、脂肪等杂质方面更有优势,故本方法选用C18柱作为净化固相萃取柱。在淋洗试剂的选用上,大部分阿维菌素类药物使用异辛烷或正己烷淋洗进一步除脂[20-25],考虑到莫昔克丁的脂溶性较强,本试验选用乙腈和30%乙腈水溶液活化C18柱,并用30%乙腈水溶液进行淋洗,在进一步有效去除杂质的同时,保证了药物的回收率(图4)。
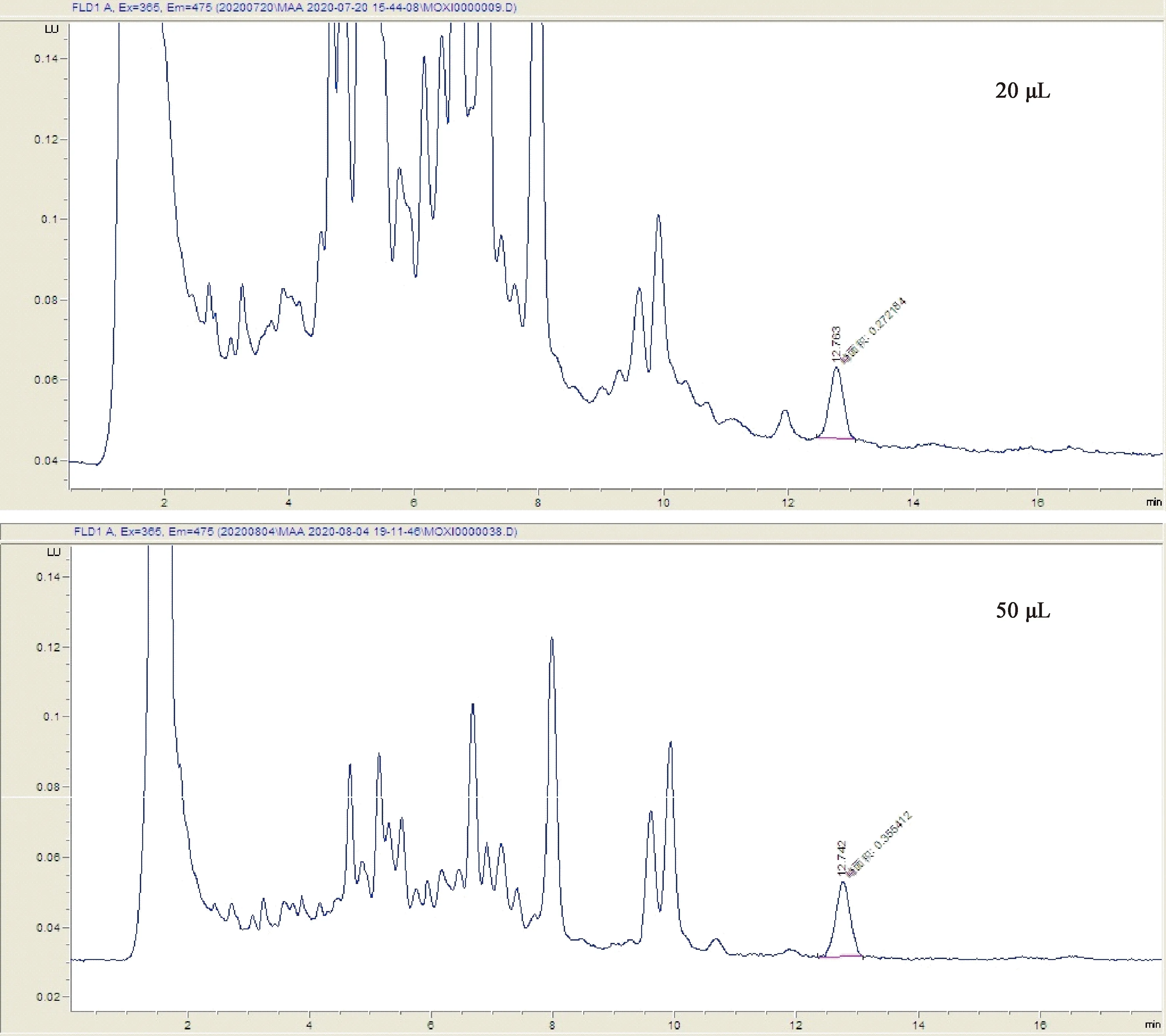
图4 提取溶剂对杂质的影响(羊肝脏)
3.3 衍生化条件的优化 阿维菌素类药物衍生化试剂的选用,普遍使用A液:N-甲基咪唑-乙腈(1+1,V/V)和B液:三氟乙酸酐-乙腈(1+2,V/V)[24-26]。本试验分别对衍生化的温度、时间、光照条件进行了摸索,发现反应若在室温下进行,则衍生化时间较长,若在加热条件下进行,衍生化速度快且充分。在反应温度和时间一定的前提下,在光照和避光两种条件下,前者药物的峰面积均明显低于后者。在衍生化试剂的选择上,本试验分别选用了不同品牌的试剂进行比对,试验发现,国药品牌与麦克林品牌N-甲基咪唑均适用于本试验,且回收率结果基本一致,但分别选用国药品牌与TCI品牌三氟乙酸酐进行衍生化后,在检测结果信噪比相同的情况下,选用TCI品牌三氟乙酸酐衍生化的样品峰高明显低于国药品牌。故本试验选用国药品牌试剂,在避光、65 ℃条件下反应15 min作为衍生化条件,并将衍生产物与甲醇混合形成醇式衍生物,具有较好稳定性,4 ℃保存24 h后进样,测得药物峰面积基本不变。
3.4 组织适用性的优化 莫昔克丁为第三代阿维菌素类药物,目前文献报道中尚且没有一种方法能同时应用于多种动物组织中莫昔克丁的残留检测[21-23]。本研究为了更好的适应我国现阶段食品安全监测工作需要,完善相应药物检测方法,利用现有检测技术手段,通过对样品的提取、净化、衍生化等条件的优化,建立了可同时适用于牛、羊的肌肉、脂肪、肝脏及肾脏样品中莫昔克丁的残留检测方法,进一步完善了我国兽药残留检测标准体系。
本试验建立了一种适用于牛、羊肌肉、肝脏、肾脏、脂肪等多种组织中莫昔克丁的高效液相色谱法。该方法操作简单、准确性好、灵敏度高、成本低,适用于莫昔克丁的分析检测。本方法的建立,进一步完善了我国兽药残留检测标准体系,为食品安全检测提供了有效技术手段。
猜你喜欢
煤化工(2022年3期)2022-07-08
日用电器(2022年3期)2022-04-14
中国土壤与肥料(2021年5期)2021-12-02
科学技术创新(2021年19期)2021-07-16
今日农业(2020年22期)2020-12-14
首都食品与医药(2020年1期)2020-10-21
食品界(2017年7期)2017-08-24
科技创新与应用(2017年1期)2017-05-11
山东工业技术(2016年10期)2016-09-06
科技与创新(2016年10期)2016-05-28
