
小儿胃畸胎瘤5例临床病理特征及预后分析
2022-03-22 14:10:20刘美莲
临床与实验病理学杂志 2022年2期
刘美莲,陈 莲
畸胎瘤(teratoma)是一种来源于生殖潜能干细胞的肿瘤,是儿科最常见的生殖细胞肿瘤,主要发生于性腺组织,发生于性腺外的主要以颅内、纵隔、腹膜后、骶尾部多见,而发生于肝脏、肾脏及胃等腹腔内脏器的畸胎瘤较为罕见。本文收集5例发生于胃部的畸胎瘤,探讨其临床病理学特征及与预后的相关性,以提高临床与病理医师的认识水平。
1 材料与方法
1.1 临床资料收集2010~2019年福建省厦门市儿童医院和复旦大学附属儿科医院经手术切除、病理诊断为胃畸胎瘤的5例标本。由两名5年以上工作经验的病理诊断医师对5例病理切片进行复核,并对患儿的临床病史、手术记录及影像学资料进行分析和总结。
1.2 方法标本均经10%中性福尔马林固定,常规脱水,石蜡包埋,4 μm厚切片,行HE及免疫组化EnVision两步法染色。抗体包括CKpan、vimentin、S-100、CD56、Syn、GFAP、Ki-67、PHXO2B及AFP等,均购自福州迈新公司;二抗试剂购自罗氏公司。具体操作步骤严格按试剂盒说明书进行,采用全自动免疫组化仪染色,设立阴阳性对照。
2 结果
2.1 临床特征本组5例胃畸胎瘤,男性3例,女性2例;年龄16天~3岁,中位年龄7个月。临床主要表现为腹胀、腹泻、腹痛,其中3例伴呕吐及贫血等症状,3例伴腹部包块,腹部CT平扫+增强示肿瘤位于胃小弯前壁2例,胃大弯后壁3例,均为囊实性占位,密度不均增高影,可见钙化高密度影,增强后均可见实性部分呈明显强化(图1),血AFP值均有升高(表1)。
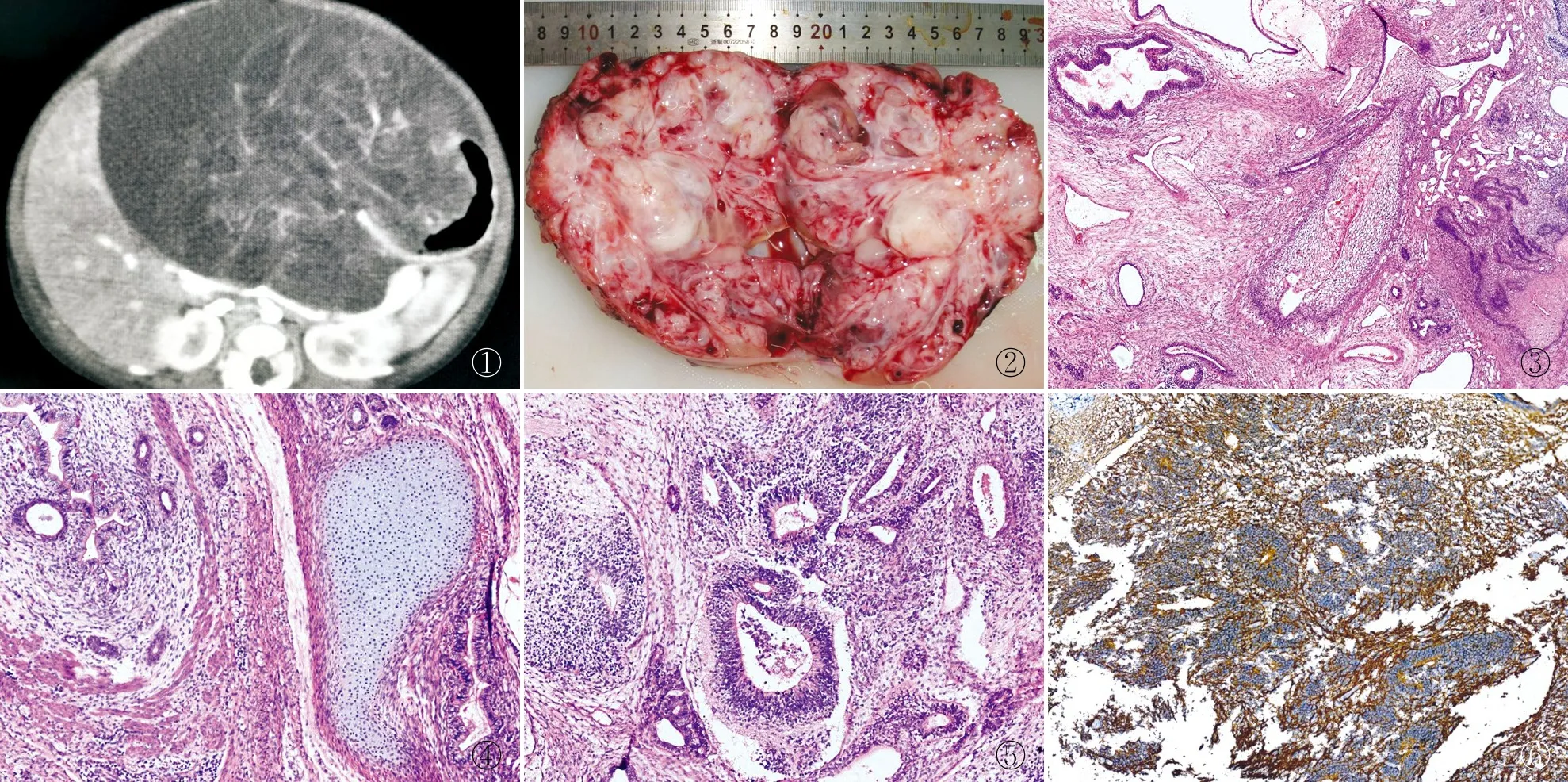
图1 CT增强示胃内囊实性肿块影,部分区呈不同程度强化 图2 肿物呈多房囊实性,囊腔内见黏液样物,实性区呈灰白色 图3 实性区见分化成熟的鳞状上皮,囊腔内衬单层肠道柱状上皮 图4 囊腔内衬单层或假复层柱状上皮,实性区见平滑肌及未成熟软骨组织 图5 组织中见多灶性原始神经管 图6 原始神经组织中CD56阳性,EnVision两步法

表1 5例胃畸胎瘤的临床资料
2.2 病理检查
2.2.1眼观 送检胃部分切除肿瘤或肿瘤单独切除标本,切开胃壁可见囊实性肿物,术中见肿瘤位于胃小弯前壁2例、胃大弯后壁3例,其中2例向胃腔外突出,3例向胃腔内突出。肿瘤大小8.0 cm×6.0 cm×5.0 cm~13.0 cm×12.0 cm×8.0 cm,有包膜,切面呈囊实性,囊性区可见黏液样物,实性区域呈灰白、灰红色(图2),部分区可见骨骼或牙齿,其中3例肿瘤表面覆盖胃黏膜组织,黏膜未见出血及溃疡形成。
2.2.2镜检 5例肿物切片中见分化成熟的鳞状上皮、肠道黏膜上皮(图3),平滑肌、纤维脂肪组织、皮肤附件、软骨(图4)、骨组织及较成熟的神经组织,其中3例见多灶性未成熟的原始神经管(图5)。
2.3 免疫表型上皮成分CK(+),间叶成分vimentin(+),神经组织(包括未成熟原始神经管)CD56(图6)和Syn均(+),分化成熟的脑神经组织S-100、GFAP(+),Ki-67增殖指数在未成熟神经管区域高表达(约15%)。
2.4 病理诊断3例为未成熟型畸胎瘤,2例为成熟型畸胎瘤。
2.5 治疗及随访5例均以手术治疗为主,其中3例行肿块联合周边胃壁切除术,2例行单纯肿块切除术,肿块切除后行胃壁缝合术,术后均未行化疗或放疗。1例失访,4例随访24~36个月,患者均预后良好,未见复发或转移。
3 讨论
畸胎瘤是来自于生殖细胞具有2个胚层以上组织衍生物的肿瘤[1],可发生于性腺内或性腺外,性腺内多见于较大的儿童,性腺外常见于新生儿及婴儿,成人也有报道[2]。发生部位常见于身体中线部位,如骶尾部、纵隔、腹膜后和中枢神经系统,其中以骶尾部、纵隔多见,而发生于胃的畸胎瘤较为罕见,被认为由胃壁的具有多能细胞产生,是由内、中、外三层原始胚层演变而来的胚胎组织残留于胃部所致[3]。胃畸胎瘤首次由Eusterman等[4]于1922年报道,其发生率不足儿童畸胎瘤的1%[5-6],好发年龄主要在1岁以内,以新生儿多见[5,7],以男性为主。本组5例胃畸胎瘤中最小年龄仅16天,最大年龄3岁,中位年龄为7个月,男女比为3 ∶2,与文献报道一致[8]。
临床症状主要表现为腹部包块、腹胀、腹痛,伴或不伴呕吐、血便及贫血等各种症状,伴或不伴血AFP值的升高,文献报道少数病例可发生于胃的自发性破裂[9]。胃畸胎瘤可发生于胃的任何部位,90%以上发生于胃大弯侧,胃的后壁多见。肿瘤发生于胃后壁常向胃外生长,临床腹部包块不明显,症状也较轻;而向胃内生长型,易引起胃的蠕动功能紊乱,从而导致胃肠梗阻。因此,临床往往可触及腹部包块并伴腹痛、呕吐等症状,影像学提示胃囊实性占位[10-11]。本组5例中2例向胃腔外生长,临床表现腹胀、腹泻,腹部包块均不明显,3例向胃腔内生长,可触及腹部包块伴腹痛、呕吐及贫血,5例影像学均提示胃囊实性占位。
胃畸胎瘤与发生性腺畸胎瘤的形态学相似,由2个或3个胚层分化的组织构成,或除了中胚层以外的单胚层组织构成的肿瘤,可能与具有生殖潜能的先天性胚胎组织残存于胃部有关[3,12]。根据WHO女性生殖系统肿瘤分类将畸胎瘤分为:(1)成熟型畸胎瘤(mature teratoma);(2)未成熟型畸胎瘤(inmature teratoma);(3)单胚层畸胎瘤。在未成熟畸胎瘤中,主要根据肿瘤组织含有未成熟原始神经管成分的多少又分为Ⅰ~Ⅲ级。未成熟原始神经管结构由复层高柱状细胞构成,细胞排列拥挤,胞质嗜碱性,细胞核致密浓染,核质比增大,核分裂象可见于管壁全层,管腔内面光滑,腔外缘不规则[13],需与高柱状细胞构成的未成熟腺体鉴别,免疫组化标记CD56呈强阳性,CK阴性,GFAP呈弱阳性或阴性。文献报道GFAP阳性是畸胎瘤成熟的有意义指标,其在成熟畸胎瘤的神经组织中呈强阳性,而在未成熟畸胎瘤的神经组织呈弱阳性[14]。CD56属于神经细胞黏附分子,主要分布于多数神经外胚层来源细胞、组织和肿瘤中[15]。本组5例肿物均呈囊实性,囊性区为黏液样物,实性区域可见毛发、脂肪及骨骼。镜下均可见肠黏膜上皮、鳞状上皮及皮肤附件,间叶组织为纤维组织、软骨、骨和脂肪组织,还可见神经组织,免疫组化标记Syn、GFAP和CD56均阳性,其中3例中见原始神经管状结构,CD56呈强阳性,GFAP呈弱阳性,提示为未成熟型畸胎瘤。
鉴别诊断:(1)原发于胃的混合性生殖细胞肿瘤:两者临床起源、组织学形态、免疫表型有相似性,但混合性生殖细胞肿瘤是具有两种或以上不同生殖细胞肿瘤成分,至少一种为原始生殖细胞,最常见为卵黄囊瘤成分,通常血清AFP异常升高,如临床出现AFP升高,切片仅为畸胎瘤成分,应多取材,仔细阅片,术后密切随访。(2)炎性肌纤维母细胞性肿瘤:其好发于儿童和青少年,以肠系膜、腹膜后及盆腔多见,部分可见于胃肠道。临床表现为胃肠道梗阻症状,大体多呈结节状或分叶状,质硬。镜下主要由梭形纤维母细胞和肌纤维母细胞构成,间质内伴大量的炎细胞浸润,免疫组化标记vimentin、SMA、MSA或desmin等阳性,约50%病例ALK阳性,2p23呈ALK基因重排。(3)胃母细胞瘤:主要见于儿童、青少年,多发于胃窦和胃体部,可累及胃壁全层,肿瘤多呈实性,部分为囊性。镜下为上皮和间叶成分不同比例混合构成,免疫表型为双向阳性,但组织学缺乏胃畸胎瘤的皮肤、骨及软骨等多胚层组织成分。(4)胃间质瘤:主要发生于成人,儿童以琥珀酸脱氢酶B(succinate dehydrogenase B, SDHB)缺陷型胃间质瘤为主,多呈结节状,瘤细胞呈上皮样或梭形,呈交织的短条束状或漩涡状排列,免疫组化标记CD34、CD117、DOG1均阳性,SDHB阴性,分子检测SDH亚单位功能丧失性胚系突变。(5)部分向胃腔外生长的胃畸胎瘤,术前需与腹膜后神经母细胞瘤、肾母细胞瘤及横纹肌肉瘤等进行鉴别诊断,由于肿瘤发生部位、病理形态及免疫表型与PHOX2B及肌源性标记等不相同,术后可明确诊断。
胃畸胎瘤以手术切除为主要治疗手段,切除的范围取决于肿瘤涉及的胃面积,手术完整切除预后良好,对于成熟型畸胎瘤和未成熟型畸胎瘤Ⅰ或Ⅱ级的肿瘤,通常不需要辅助放、化疗[16-17]。本组4例进行随访,时间24~36个月,其中4例均预后良好,1例失访。
综上所述,小儿胃畸胎瘤罕见,临床症状与肿瘤发生部位密切相关,尽管特征性的影像学征象可帮助诊断,但最终确诊仍依靠病理检查。目前,治疗手段以肿瘤完整切除为主,多数患者无需辅助化疗或放疗,绝大多数患者预后良好。患者术后应长期随访行影像学检查,且需监测血清AFP的变化。
猜你喜欢
文萃报·周五版(2022年24期)2022-06-21 20:55:40
中国临床医学影像杂志(2021年5期)2021-08-13 09:01:34
中国CT和MRI杂志(2018年12期)2018-12-13 08:01:28
哈尔滨医药(2016年1期)2017-01-15 13:43:16
中国男科学杂志(2016年5期)2016-12-01 05:20:22
中国继续医学教育(2015年5期)2016-01-07 07:38:16
山东医药(2015年14期)2015-04-04 14:00:07
食品工业科技(2014年15期)2014-03-11 18:17:46
微创泌尿外科杂志(2014年4期)2014-02-28 17:28:44
中国医学影像学杂志(2012年3期)2012-12-08 06:15:18
