
抑制TAK1可加重甲苯二异氰酸酯诱导的哮喘小鼠气道炎症
2022-03-20 03:28:44杨淑銮赵文驱彭显如蓝紫涵黄俊文韩慧珊蔡绍曦赵海金
南方医科大学学报 2022年2期
哮喘具有多种临床表型和内因型,大部分哮喘患者在吸入性糖皮质激素等治疗后可以有效控制,但仍有5%~20%的哮喘患者即使用高剂量吸入性糖皮质激素治疗仍控制不佳,这些重度哮喘或激素抵抗型哮喘的发生机制尚未完全阐明。其中,丝裂原活化蛋白激酶(MAPK)信号通路的过度激活可能在激素抵抗过程发挥着重要作用。
本课题组长期关注甲苯二异氰酸酯(TDI)哮喘模型气道炎症发生机制,有研究发现,TDI诱导的是一种激素抵抗型哮喘模型,我们前期研究发现MAPK信号通路在TDI哮喘模型明显活化,且阻断MAPK信号通路能有效减轻TDI哮喘小鼠的气道炎症、气道屏障破坏及气道高反应性,提示MAPK通路可能与激素抵抗有关。转化生长因子β激活激酶1(TAK1)是激活下游包括NF-κB和MAPK信号通路活化的关键酶,在调节各种细胞类型的炎症、免疫和细胞死亡发挥关键作用,为治疗炎症性疾病的潜在靶点。有小样本研究证实阻断TAK1可以恢复激素抵抗哮喘患者来源的巨噬细胞对激素的敏感性。有文献报道TAK1可能通过影响Th17通路在中性粒细胞哮喘免疫调节中发挥重要作用。然而,TAK1是否在TDI诱导的激素抵抗型哮喘模型扮演重要角色及如何发挥作用尚不清楚。因此,本研究旨在探讨TAK1对TDI哮喘小鼠气道炎症的影响。
1 材料和方法
1.1 试剂
丙酮、橄榄油、TDI(sigma);TAK1 抑制剂5Z-7-Oxozeaenol(sigma),RIPK1抑制剂Necrostatin-1(MCE),CCK8(同仁),p-TAK1、TAK1、p-ERK、P38、p-P38、JNK、p-JNK、RIPK1、p-RIPK1 等一抗(CST),ERK 一抗(proteintech),Caspase8 一抗(proteintech),GADPH 一抗(proteintech),IRDye荧光二抗(licor),ELISA试剂盒(赛默飞),苏木素染液,伊红染液,凯基全蛋白提取试剂盒等。
1.2 TDI哮喘模型建立及动物干预
SPF级雄性BALB/c小鼠64只(南方医科大学实验动物中心),6~8周,本实验经南方医院实验动物伦理委员会批准。
对应各部分新增传感器等设备,一方面,对原PLC控制柜进行改造,增加水冷系统等部分的数据采集和处理模块;另一方面,对控制芯片进行重新编程,拓展其监测范围和功能。其次,在柜体内增加以太网模块,保证PLC控制层与上机位的数据通信,从而为现场设备的远程监控提供基础。通过对PLC控制柜的改造,其主要结构和功能如图3,PLC芯片可对传感器的数字和模拟信号进行处理,并按照预定程序,向继电器发出动作信号,同时控制指示灯状态。另外,及时将各种数据和处理结果经由交换机传输至上机位。
TDI哮喘模型构建:于造模第1、8天用0.3%TDI通过耳背皮肤致敏小鼠,第15、18、21天通过雾化吸入3%TDI激发小鼠,TDI溶于丙酮和橄榄油的混合物(AOO)中,空白对照小鼠只接受AOO致敏和激发,具体剂量参考先前研究。
与本研究抑制TAK1促炎作用相一致,在内毒素(LPS)诱导的小鼠模型中,与野生型相比,TAK1敲除小鼠的肺部炎症明显加重,伴IL-1β、IL-6、TNF-α水平明显增加,作者同时发现p38和JNK激酶的磷酸化以及活性氧ROS水平明显增加,进一步发现TAK1敲除小鼠来源的中性粒细胞IL-1β、IL-6、TNF-αmRNA表达明显增加。提示TAK1在不同的模型中发挥着不同的调节作用。
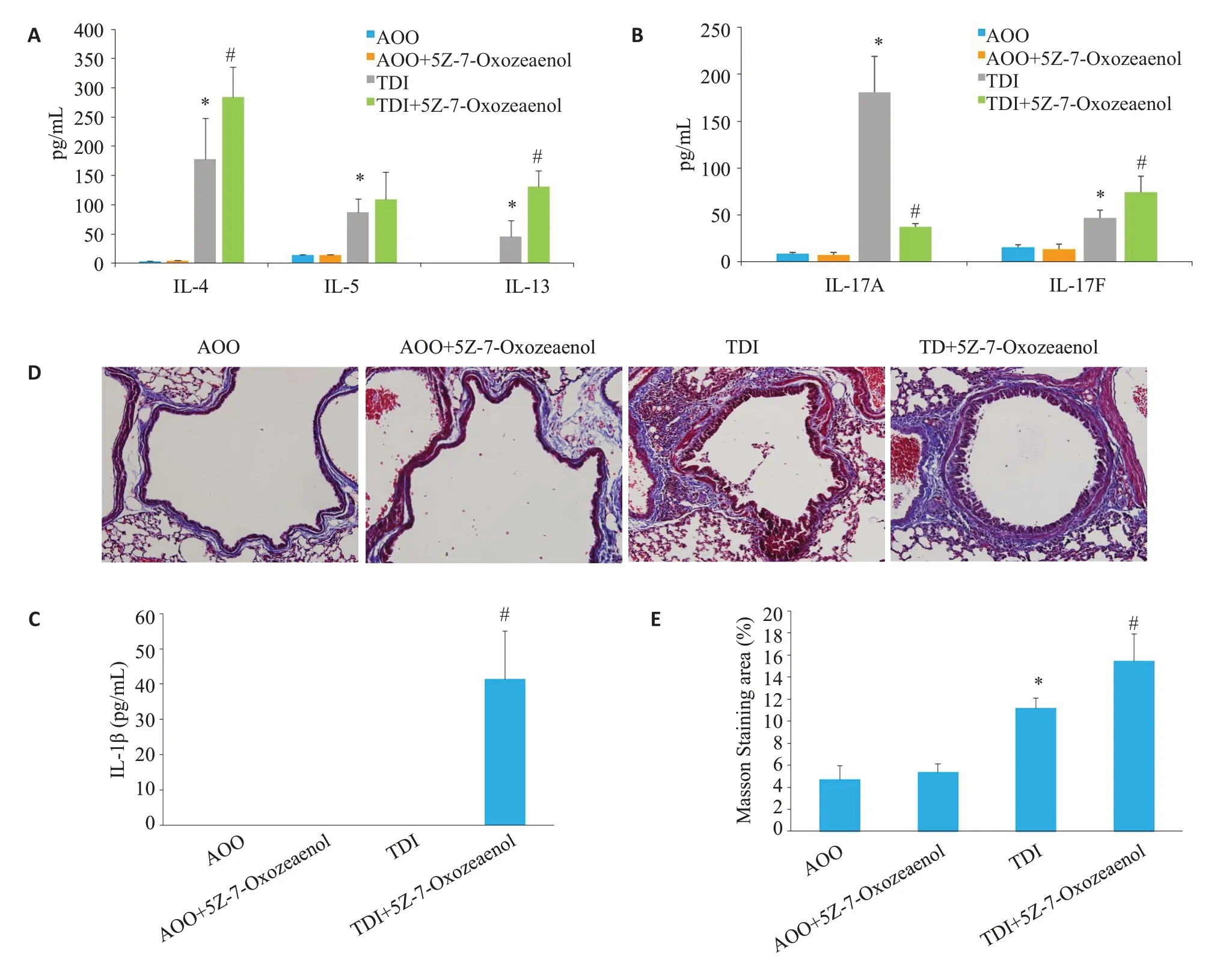
TDI致敏及激发建立了以中性粒细胞为主的混合粒细胞性气道炎症的哮喘模型(图2E),同时伴有气道高反应性及气道重塑(图2A,图3D)。使用5Z-7-Oxozeaenol,进一步加重了小鼠的气道高反应性及增加了血清IgE浓度(<0.05,图2A、B)。组织切片HE染色显示,阻断TAK1进一步加重了气道周围炎症细胞增多,上皮增生;进一步评估小鼠气道重塑指标,结果显示,TDI能显著引起小鼠气道粘液分泌的增加及上皮下胶原沉积(<0.05,图3D、E)。PAS 染色显示,阻断TAK1 对于粘液分泌没有明显影响(=0.850),MASSON染色则显示其能明显加重哮喘小鼠的胶原沉积(<0.05);而单独使用TAK1抑制剂,对对照组的气道炎症(=0.147)、粘液分泌(=0.85)及胶原下沉积(=0.63)没有明显影响。
进一步探讨RIPK1在体内模型的作用,将另外32只小鼠随机分为4组,8只/组:AOO组,TDI组,TDI+5Z-7-Oxozeaenol组,TDI+5Z-7-Oxozeaenol+Necrostatin-1组。TDI+5Z-7-Oxozeaenol 组、TDI+5Z-7-Oxozeaenol+Necrostatin-1 组每次激发3 h 前腹腔注射5 mg/kg TAK1抑制剂(5Z-7-Oxozeaenol)和/或RIPK1活性抑制剂(Necrostatin-1,5 mg/kg),两种抑制剂给药时间间隔30 min。AOO组、TDI组给予同等剂量的DMSO。
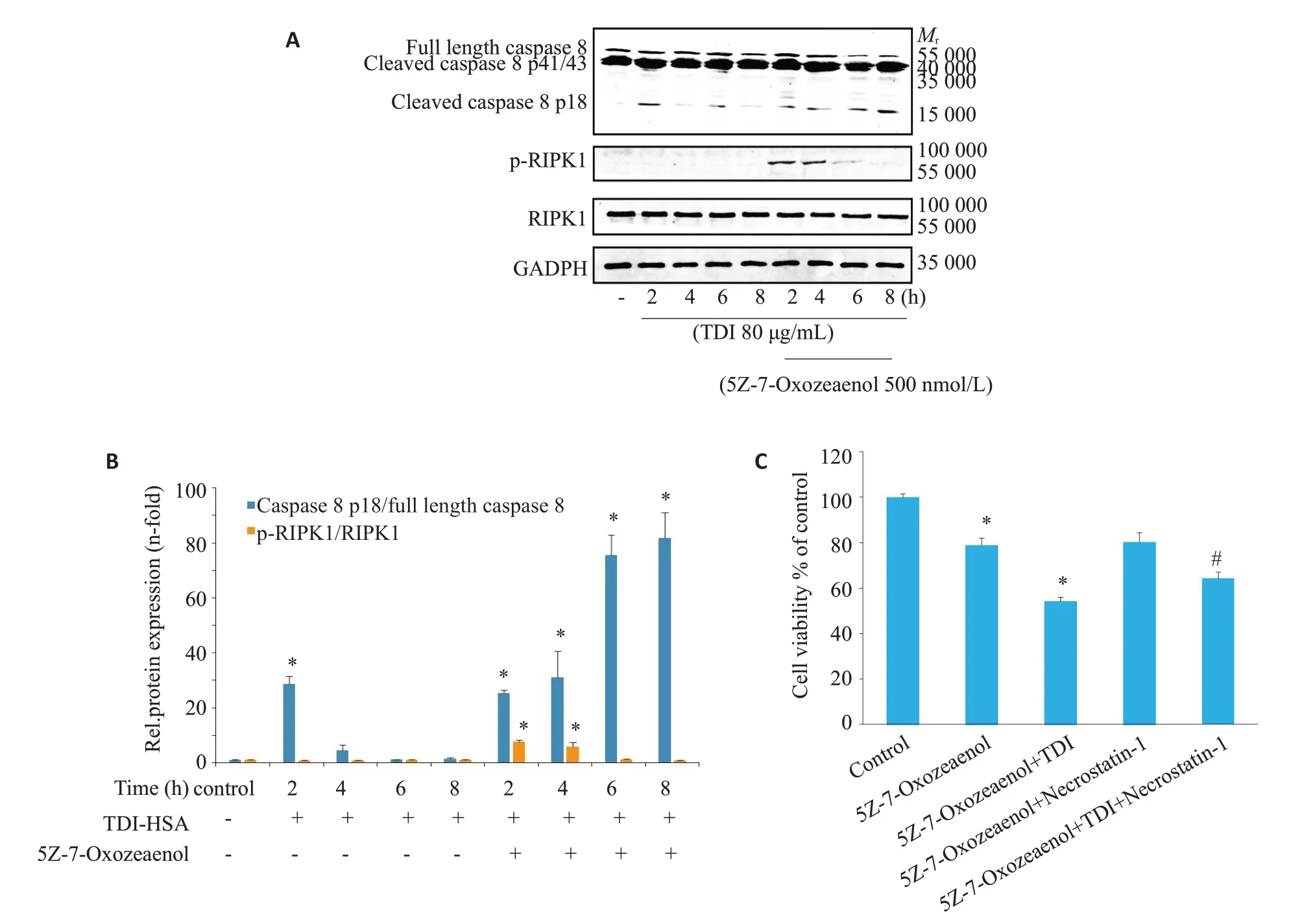
Western blotting结果显示,TDI-HSA刺激与TDIHSA联合TAK1抑制剂共同处理,未能明显影响RIPK1蛋白水平,而TDI-HSA 联合TAK1 抑制剂,在2 h 时RIPK1磷酸化水平上升(<0.05),随后降低。单独使用TDI-HSA 刺激RAW264.7,Caspase 8 在2 h 出现活化(<0.05),随后快速降低,而TDI-HSA联合TAK1抑制剂,其Caspase 8活性明显升高,且随后进一步升高(<0.05)。进一步使用RIPK1活性抑制剂Necrostatin-1,明显恢复TDI-HSA联合TAK1抑制剂引起的细胞活力的下降(<0.05),而对于单独TAK1抑制剂引起的细胞活力水平的下降没有明显影响(=0.48)。
金叶风箱果为蔷薇科风箱果属落叶灌木,高可达3 m,株型呈拱形多分枝,小枝光滑无毛,冠形开展。原产北美,在我国大连、北京、重庆等多个城市广泛栽培。金叶风箱果整个生长季新梢1~8片叶一直是金黄色。叶片春季和初夏金黄色,中夏至秋季黄绿色,秋末叶呈黄红相间色[1]。夏季花序密集,花色淡雅,入秋果实变红,可用于绿篱、假山旁、路旁、海岸种植,是优良的观叶、观花、观果植物[2]。但其优良性状在生产上一般通过扦插繁殖得以保持下来,为了给培育优质壮苗提供科学依据,进行了金叶风箱果的扦插繁殖试验。
1.3 RAW264.7培养,刺激及细胞活力检测。
小鼠单核巨噬细胞株(RAW264.7,中科院),其采用含10%胎牛血清的DMEM培养基,在37 ℃、5%CO培养箱中传代培养,细胞生长状态良好时用于实验,予不含血清的DMEM培养细胞12 h,即饥饿处理后,予不同浓度5Z-7-Oxozeaenol、1 μmol/L Necrostatin-1 及80 μg/mL TDI-HSA刺激细胞一定时间,TDI-HSA复合物按文献配置。细胞活力依照CCK8试剂盒说明书进行检测。
1.5 Western blot
采用凯基全蛋白提取试剂盒抽提细胞总蛋白,肺组织总蛋白,BCA法测定蛋白浓度,等量蛋白上样,使用BIO-RAD进行电泳及转膜,5%BSA封闭1 h,一抗4 ℃过夜,TBST洗膜30 min,二抗孵育1 h,LI-COR红外荧光扫描成像检测。
1.6 统计学分析
TDI哮喘小鼠Th2/Th17炎症因子升高(<0.05,图3A、B),给予TAK1抑制剂增加了IL-4及IL-13水平(<0.05),而IL-5差异无统计学意义(=0.21)。阻断TAK1升高了TDI哮喘小鼠淋巴上清IL-17F的水平(<0.05),降低了TDI引起IL-17A的水平的升高(<0.05)。同时,单纯的TDI致敏及激发组,未能在BALF中检测到IL-1β,而给予TAK1抑制剂,检测到IL-1β升高(<0.05,图3C)。
2 结果
2.1 TAK1抑制剂降低TDI诱导的TAK1磷酸化水平的升高及MAPK信号通路的活化
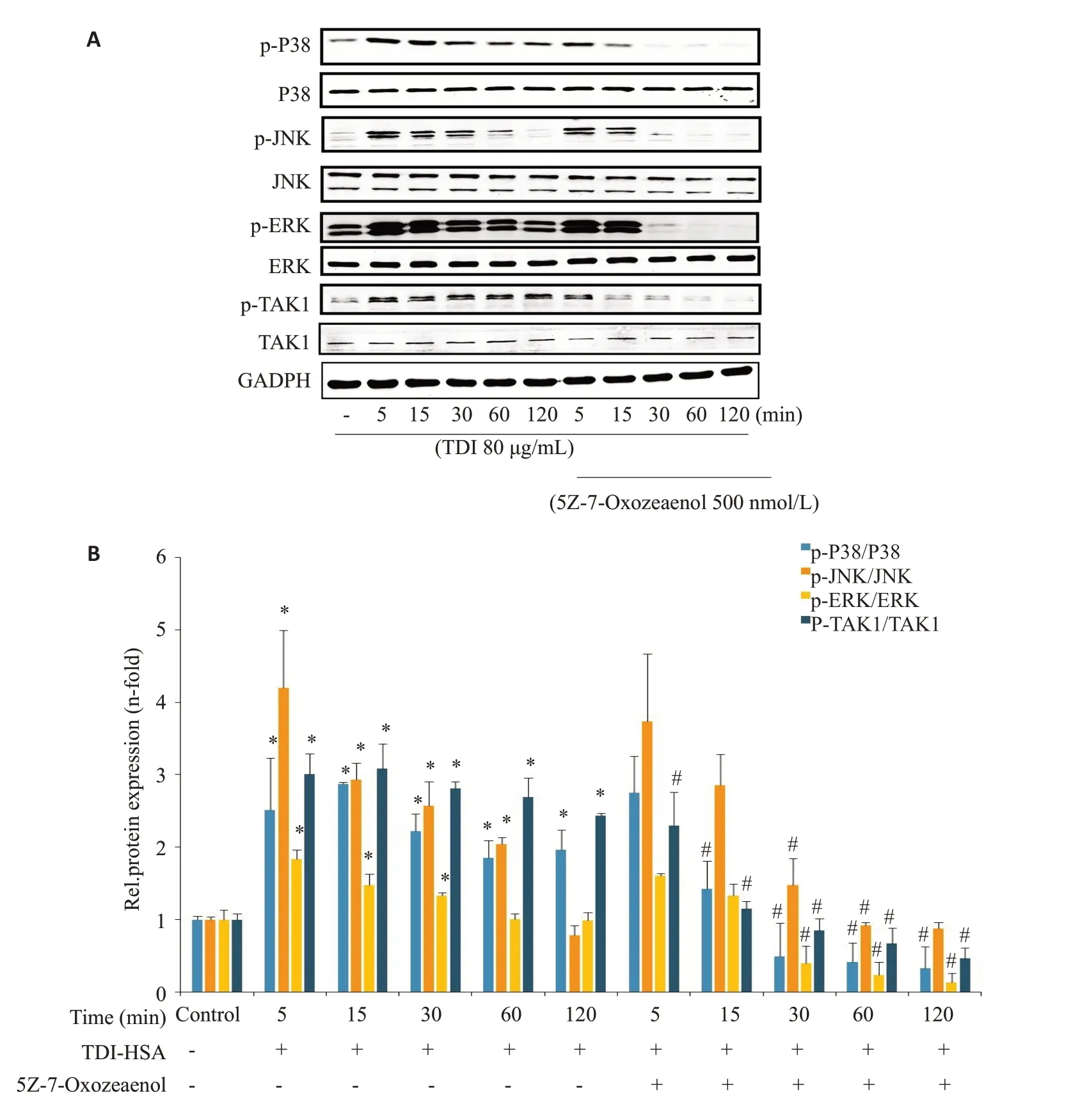
结果显示,阻断TAK1不仅降低了TDI引起的肺组织TAK1磷酸化水平的升高,同时降低了其下游分子P38 MAPK,JNK及ERK的磷酸化水平(<0.05,图1)。

2.2 阻断TAK1进一步加重了TDI诱导的气道高反应性及气道炎症
取pH=6.0的枸杞蛋白质溶液8.0ml,加入相对应的(NH4)2SO4固体[11],使蛋白质溶液的(NH4)2SO4饱和度达20%,摇匀,静置3~4h后以8000r/min离心10min,倾出上清液并测量体积,重复以上步骤,直到蛋白质溶液的(NH4)2SO4饱和度达100%。得到(NH4)2SO4饱和度分别为20%、40%、60%、80%、100%的枸杞蛋白沉淀。向5次离心后的倾出上清液的离心管中加入0.2mol/l pH=6.0的PBS缓冲液5.0ml,轻轻摇动,使贴于管壁上的蛋白质溶解后置于冰箱中保存。
2.3 阻断TAK1增加TDI哮喘小鼠淋巴上清Th2/Th17炎症水平
采用SPSS 20.0统计软件分析处理,定量资料以均数±标准差表示,组间比较采用单因素方差分析;符合方差齐性检验者,组内比较使用LSD方法检验,不符合方差齐性检验者,组内比较使用Dunnett's T3分析,<0.05时认为差异具有统计学意义。
观察组组织中COX-2和VEGF表达的阳性率均显著高于对照组(P<0.001),LMP1的阳性率明显低于对照组(P<0.001),另外观察组血清中IL-8的表达水平较对照组明显增高(P<0.001),见表1。
2.4 TDI-HSA 联合TAK1 抑制剂对巨噬细胞RAW264.7活力的影响
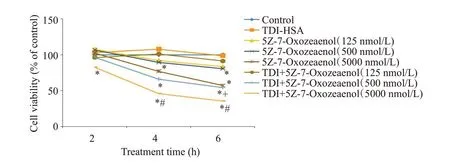
TAK1抑制剂可以降低TDI哮喘小鼠肺泡灌洗液(BALF)分类计数细胞中巨噬细胞的比值(图2D)。单独使用TDI-HSA(80 μg/mL)刺激细胞,未降低细胞活力(图4)。而单独使用TAK1抑制剂,可以在较高浓度时(5000 nmol/L)时,在早期(2 h)时细胞活力降低(<0.05),而较低浓度(500 nmol/L)时到6 h时细胞活力出现降低(<0.05)。同时,与单独使用TAK1 抑制剂相比,与TDI-HSA 联合使用降低了细胞活力(<0.05)。为探讨抑制TAK1加重TDI诱导哮喘小鼠气道炎症的机制,以下实验中,选用500 nmol/L的抑制剂浓度处理细胞。

2.5 TAK1抑制剂对TDI-HSA诱导的RAW264.7 TAK1磷酸化水平的影响
本研究首次发现在TDI哮喘模型中,抑制TAK1进一步加重了TDI哮喘小鼠的气道炎症,其机制可能与促进巨噬细胞RIPK1介导的细胞死亡有关。有研究报道,在小鼠颗粒物诱导的原代巨噬细胞模型中,敲除TAK1可以降低IL-6、IL-33的表达,与野生组型比,肺巨噬细胞TAK1条件敲除小鼠肺部炎症指标及相关细胞因子表达明显降低。本课题组既往的研究发现P38 MAPK、JNK、ERK等MAPK信号通路在TDI哮喘模型中明显活化,阻断JNK 能明显减轻气道炎症。TAK1作为MAPK信号通路的上游激酶,因此,我们设想TDI 哮喘小鼠的气道炎症能被TAK1 抑制剂所减轻。结果发现,TAK1抑制剂确实可以显著抑制TDI诱导的MAPK信号通路的活化。然而,非常有趣的是,TAK1抑制剂却进一步加重了TDI哮喘小鼠的气道炎症,上皮的脱落,气道的高反应性。这提示,TDI哮喘模型中,TAK1抑制剂加重哮喘气道炎症并不是通过促进下游的MAPK信号通路活化而发生。
2.6 TAK1抑制剂对TDI-HSA诱导的MAPK信号通路活化的影响
TDI-HSA 刺激RAW264.7 细胞,5 min 即引起p-P38、p-JNK 及p-ERK 等蛋白表达水平明显升高(<0.05)。而给予TAK1 抑制剂与TDI-HSA 共同处理RAW264.7、JNK、P38及ERK蛋白的磷酸化水平都降低(<0.05)。
2.7 阻断TAK1促进了RIPK1介导的细胞死亡
按照上述方法造模后,于第22天检测气道反应性后,处死小鼠,取右肺进行石蜡包埋,常规脱蜡至水进行HE染色、PAS染色及MASSON染色。气道反应性及淋巴细胞培养详见文献[12]。
2.8 使用RIPK1抑制剂可以恢复TAK1抑制剂引起的TDI哮喘小鼠气道炎症及气道重塑的加重
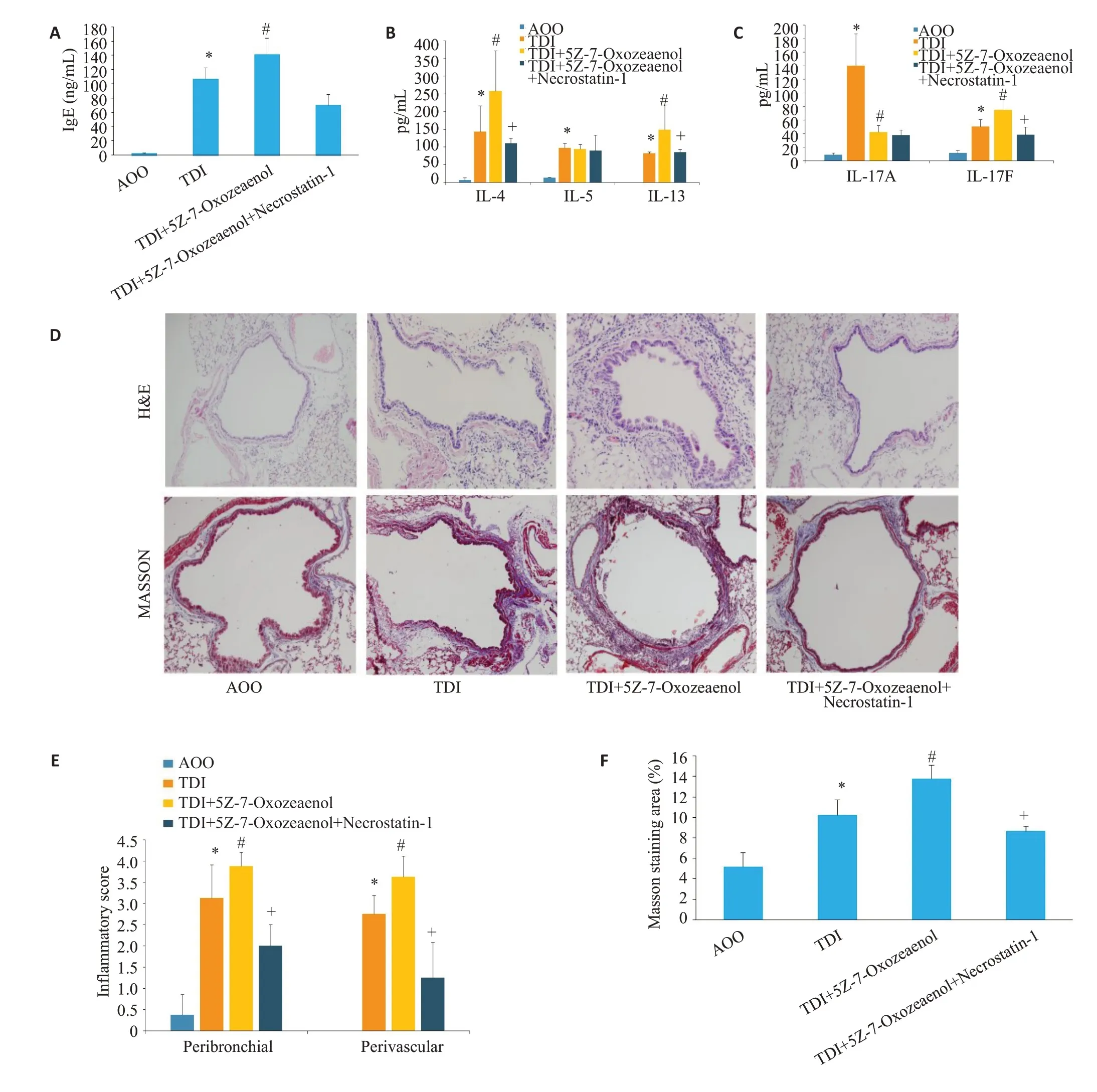
与单独使用TAK1抑制剂对TDI哮喘小鼠相比,联合使用RIPK1 活性抑制剂可减轻气道炎症(<0.05,图7),降低血清IgE浓度及淋巴上清中IL-4、IL-13及IL-17F水平及改善气道重塑(<0.05)。
渡槽为现浇肋拱模架,盘扣模架底座搭设在横桥向工字钢上;盘扣支架的横向剪刀撑应与支架底部横向工字钢进行焊接,将工字钢、贝雷梁与钢支墩之间用套箍进行连接;下部钢管柱采用钢筋混凝土条形基础,并预埋钢板埋件。
3 讨论
检测各组间TAK1水平及其磷酸化程度显示,单独TDI-HSA刺激RAW264.7后,TAK1的磷酸化水平在短时间内即升高(<0.05,图5)。而使用TAK1抑制剂5Z-7-Oxozeaenol(500 nmol/L),可以降低TDI-HSA引起的TAK1磷酸化水平的升高(<0.05)。
为探讨TAK1在TDI哮喘模型中的作用,将32只小鼠随机分为4 组,8 只/组:AOO 组,AOO+5Z-7-Oxozeaenol 组,TDI 组,TDI+5Z-7-Oxozeaenol 组。AOO+5Z-7-Oxozeaenol 组、TDI+5Z-7-Oxozeaenol 组于每次激发3 h前腹腔注射5 mg/kg TAK1抑制剂(5Z-7-Oxozeaenol)干预TAK1,AOO组、TDI组给予同等剂量的DMSO。
巨噬细胞是肺部最重要的免疫细胞之一,在变应原诱发的哮喘气道炎症中发挥重要作用,巨噬细胞可以通过分泌多种炎症介质,包括IL-1β、IL-33、TNF-α等在哮喘的发生发展中发挥着重要的作用,亦有研究发现,TDI哮喘中巨噬细胞活化。因此,本研究假设在TDI哮喘模型中,TAK1抑制剂通过增加巨噬细胞的死亡,释放炎症介质,进一步加重TDI哮喘小鼠的气道炎症。与预期一致,本研究显示抑制TAK1后明显降低哮喘模型BALF中巨噬细胞比例,体外亦降低RAW264.7细胞活力,且在与TDI-HSA共同刺激时其细胞活力进一步下降,但其机制有待进一步阐明。
TAK1作为MAPK与NF-κB信号通路的上游激酶,通常参与促生存信号,研究发现当其被抑制或缺失时,细胞将发生PANoptosis,细胞从促生存信号向促死亡信号方向转变。PANoptosis包括焦亡、凋亡和坏死三种细胞死亡形式,是由含有RIPK1、ASC和caspase 8的PANoptosome的形成触发的,其中RIPK1激酶活性对驱动焦亡、凋亡和坏死混合细胞死亡表型至关重要。本研究发现,联合使用TAK1抑制剂及TDI-HSA 可以激活RIPK1 的活性,同时持续裂解Caspase 8,促进巨噬细胞的死亡,使用RIPK1抑制剂可以降低RAW264.7的死亡,同时,体内实验也证实使用RIPK1抑制剂可以显著减轻TAK1抑制剂对于TDI哮喘小鼠气道炎症的加重。与本研究一致,近期有研究发现,衰老导致TAK1表达减少会导致RIPK1驱动的神经退行性变。有研究提出耶尔森氏菌诱导细胞死亡模型,即部分巨噬细胞被耶尔森菌感染,当宿主细胞感知到TAK1被抑制时,形成由RIPK1、Caspase 8和FADD组成的细胞死亡复合体,导致细胞膜通透性增加和随后的细胞死亡,释放IL-1β,K+等,激活部分TAK1未被抑制的巨噬细胞,以促发更为迅速的炎症瀑布反应。本研究设想在TDI哮喘模型中,TAK1磷酸化水平的增高,抑制了RIPK1活性,进而限制了Caspase 8的持续活化,从而降低了巨噬细胞的死亡。然而,在TAK1抑制剂处理的哮喘模型中,是否也发生了PANoptosis,其细胞死亡是否也为包括焦亡、凋亡和坏死多种细胞死亡形式的混合细胞死亡表型,需要进一步实验验证。
心电图作图是一项比较简单的操作,但日常工作中有可能忙中出错,大批量体检、心电远程会诊中,读图者与作图者分离,读图者无法了解作图时的导联连接情况,即使初步判断有可能发生导联错接,也需要重新作图方能验证,并出具正确的报告,以便临床下一步诊治。实际情况是,患者若了解是由于作图者的失误而要求自已再来诊室重新作图,其理解及配合度不会很高。利用本文中导联错接的判断及调整方法能实现“一键还原”,既不麻烦患者,也不影响诊断的效果,值得临床推广。
本研究发现,与其他炎症性疾病不同,抑制TAK1出乎意料地加重了TDI哮喘小鼠的气道炎症,进一步发现其对于巨噬细胞死亡的影响。在不同细胞死亡途径之间存在着许多的联系,在特定刺激下,参与细胞焦亡、细胞凋亡和/或坏死的分子可以同时被激活。未来对细胞死亡之间各种联系和调节机制的深入研究,将进一步帮助理解其中的作用途径。
综上所述,在TDI哮喘小鼠模型中,TAK1阻断进一步加重了气道炎症,其机制可能与促进RIPK1信号通路及Caspase 8的持续活化,从而促发巨噬细胞的持续死亡,增加炎症介质的释放有关。
由于对排风冷量进行了回收对新风进行预冷,同时采用排风蒸发冷却技术降低了制冷系统的冷凝压力,提升了双冷源新风机组的制冷效率,制冷性能系数和除湿性能系数都比常规冷冻除湿有了显著提高。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
天津医科大学学报(2019年6期)2019-08-13 07:04:42
上海农业学报(2017年3期)2017-04-10 12:39:26
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
遗传(2014年3期)2014-02-28 20:59:01
无机化学学报(2014年10期)2014-02-28 17:33:13
金属矿山(2013年11期)2013-03-11 16:55:05
