
Neuroinflammation as a mechanism linking hypertension with the increased risk of Alzheimer’s disease
2022-03-19 03:30EktaBajwaAndisKlegeris
中国神经再生研究(英文版) 2022年11期
Ekta Bajwa, Andis Klegeris
Abstract Alzheimer’s disease, the most common type of dementia among older adults, currently cannot be prevented or effectively treated.Only a very small percentage of Alzheimer’s disease cases have an established genetic cause.The majority of Alzheimer’s disease cases lack a clear causative event, but several modifiable factors have been associated with an increased risk of this disease.Persistent midlife hypertension is one such risk factor, which can be effectively controlled through changes in diet, lifestyle, and antihypertensive drugs.Identifying molecular mechanisms linking modifiable risk factors with the increased risk of Alzheimer’s disease could enhance our understanding of this disease and lead to identification of novel targets and therapeutic approaches for effective treatments.Glial cell-driven neuroinflammation is one of the key pathological features of Alzheimer’s disease.In this review, we illustrate that neuroinflammation could also be one of the possible mechanisms linking hypertension and Alzheimer’s disease.Animal studies have demonstrated that chronically elevated blood pressure leads to adverse glial activation and increased brain inflammatory mediators.We highlight damage to cerebral microvasculature and locally activated renin-angiotensin system as the key pathogenetic mechanisms linking hypertension to neuroinflammation and the accompanying neurodegeneration.The role of tumor necrosis factor-α and interleukin-1β as pro-inflammatory signaling molecules providing this link is discussed.We also summarize the available experimental data indicating that neuroinflammatory changes and glial activation can be reversed by several different classes of antihypertensive medicines.These studies suggest antihypertensives could be beneficial in Alzheimer’s disease not only due to their ability to control the blood pressure, but also due to their antineuroinflammatory effects.Confirmation of these observations in human subjects is required and recent advances in the brain imaging techniques allowing visualization of both microglia and astrocyte activation will be essential for this research.
Key Words: Alzheimer’s disease; antihypertensive medicines; astrocytes; blood-brain barrier; high blood pressure;microglia; neurodegenerative disorders; paraventricular nucleus; renin-angiotensin system
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder that is one of the most common types of dementia worldwide.According to the Alzheimer’s Disease International, there were 46 million people living with dementia globally in 2015 and this number is projected to reach over 131 million by 2050 (Alzheimer’s Disease International, 2015).In AD, the hippocampus and frontal cortex are predominantly affected, resulting in cognitive symptoms,such as memory loss and language difficulties.The central nervous system(CNS) neuropathology of AD is characterized by the deposition of amyloid-β plaques and the formation of neurofibrillary tangles.In addition to being directly neurotoxic, these pathological structures also trigger acute neuroinflammation, which contributes to the AD pathogenesis (reviewed by Calabro et al., 2021; Scheltens et al., 2021).
Numerous studies have confirmed that lifestyle-related conditions, such as diet, hypertension, and type 2 diabetes mellitus (T2DM), increase the risk of dementia and AD (Barnes and Yaffe, 2011; Elias and Davey, 2012; Livingston et al., 2020).Some of these risk factors, such as hypertension, have wellcharacterized direct mechanistic links to the pathogenesis of dementia and AD that do not involve inflammatory processes (see Desideri and Bocale,2020).Furthermore, many of the risk factors are inter-related.For example,obesity can lead to hypertension and T2DM (Jiang et al., 2016); therefore,these three pathologies often co-occur (Barnes and Yaffe, 2011).
This article considers the inflammatory mechanisms which are part of the pathophysiology of obesity, hypertension, and T2DM.It has been proposed that the peripheral inflammation occurring in these disease processes may contribute to the neuroinflammation observed in AD brains and thereby increase the risk of developing AD (reviewed by McKenzie et al.,2017; Newcombe et al., 2018).Several different routes of transduction of inflammatory signals from the periphery to the CNS have been described,including transfer of immune mediators across the blood-brain barrier(BBB) and stimulation of vagus nerve afferents by inflammatory mediators.Furthermore, some inflammatory mediators, such as tumor necrosis factor-α (TNF)-related apoptosis inducing ligand, regulate inflammatory processes in both the periphery and CNS, representing good therapeutic targets for reducing neuroinflammation (reviewed by Di Benedetto et al.,2019; Burgaletto et al., 2020; Hashioka et al., 2021).However, control of the modifiable risk factors with pharmacological interventions or behavior modification may also represent an effective strategy for decreasing the incidence of AD or inhibiting disease progression.This is imperative since there is currently no effective treatment for this devastating disorder.A recent meta-analysis, including 12 trials with 92,135 participants, illustrates that lowering blood pressure with antihypertensive agents is associated with significantly reduced risk of cognitive impairment and dementia; however,there is no significant association between lowering of blood pressure and a change in cognitive test scores (Hughes et al., 2020).This study complements four earlier meta-analyses all demonstrating blood pressure medications lower risk of any dementia or specifically AD (reviewed by Livingston et al., 2020).Blood pressure in the studies considered by these meta-analyses is controlled by medications from different classes, including diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II(Ang-II) receptor blockers, beta-blockers, calcium channel blockers, or a combination of drugs from different classes.Interestingly, the meta-analysis by Peters et al.(2020), considering 27 studies with over 50,000 participants,demonstrates no significant differences in association between specific class of antihypertensive drugs and the incidence of cognitive decline or dementia among subjects aged 65 or older.The exception is diuretics, the use of which is associated with a reduced risk in some studies; however, this effect is not consistent across all parameters assessed (outcome, follow-uptime, and comparator group).The aim of this article is to highlight the current evidence implicating inflammation as one of the pathological pathways linking hypertension and AD.
Search Strategy and Selection Criteria
Studies published between 1997 and 2021 are cited in this review with more than half of these articles published during the last five years.The initial search was performed within OVID Medline (PubMed) databases using the following keywords: Alzheimer’s disease, neuroinflammation, hypertension,renin angiotensin system, microglia, astrocytes, and glia.Abstracts of the retrieved research articles were screened and full versions of relevant articles acquired.Further pertinent articles were identified by reviewing the reference lists of cited publications.
Neuroinflammation in Alzheimer’s Disease
Neuroinflammation represents an essential physiological response required for the removal of infectious agents and CNS repair after injury.In AD,the acute inflammatory response is triggered by increased deposition of disease-specific pathological structures including amyloid-β plaques and neurofibrillary tangles as well as by degenerating and dying cells releasing damage-associated molecular patterns.These molecules are sensed by the two main glial cell types, astrocytes and microglia, which orchestrate neuroimmune reactions in the brain (reviewed by Akiyama et al., 2000;Bachiller et al., 2018; Bajwa et al., 2019; Calabro et al., 2021).Astrocytes participate in a broad range of physiological processes, including regulation of the cerebral blood flow and permeability of the BBB, clearance of neurotransmitters and regulation of synaptic activity.Astrocytes become reactive in response to pathological structures and cooperate with microglia during neuroimmune responses (reviewed by Arranz and De Strooper, 2019;Khakh and Deneen, 2019; Trujillo-Estrada et al., 2019; Escartin et al., 2021).Microglia, the resident macrophages of the CNS, provide immune defense and also support physiological processes by removing cell debris, secreting neurotrophic factors and assisting with the formation and elimination of synapses (Gomez Perdiguero et al., 2015; Prinz et al., 2019).Microglia display ramified morphology and their processes constantly extend and retract,surveying the brain parenchyma.When microglia encounter pathogens or abnormal molecular structures, they become reactive and launch an innate immune response.Microglia can also serve as antigen-presenting cells to initiate an adaptive immune response in certain brain pathologies,including AD (Nimmerjahn et al., 2005; Schetters et al., 2017; Prinz et al.,2019).Reactive microglia are capable of secreting a diverse set of neurotoxic molecules, such as reactive oxygen and nitrogen species, L-glutamate,cathepsins, and matrix metalloproteinases (reviewed by Lindhout et al.,2021).Furthermore, the immune signaling molecules released by adversely activated microglia (e.g., cytokines, chemokines, and complement proteins)stimulate astrocytes, which aid microglia in recruiting peripheral monocytes and T-cells to the brain parenchyma, causing a sustained state of chronic neuroinflammation.Toxins released by reactive microglia, astrocytes, and peripheral immune cells induce further death of neurons, causing them to release a wide variety of damage-associated molecular patterns into the extracellular space.This leads to persistent activation of both microglia and astrocytes, thus establishing the vicious cycle of neuroimmune activation in AD (reviewed by Akiyama et al., 2000; Liddelow and Barres, 2017; Bachiller et al., 2018; Fakhoury, 2018; Bajwa et al., 2019; Burgaletto et al., 2020; Calabro et al., 2021; Scheltens et al., 2021).
Hypertension and Alzheimer’s Disease
Midlife (45-65 years) hypertension is a known contributor to the etiology of late-life (older than 65 years) dementia and is increasingly implicated as a risk factor for the development of AD (reviewed by Livingston, 2020).For example, a recent epidemiological study found that midlife stage 1 (systolic blood pressure > 140 mmHg) and stage 2 (systolic blood pressure > 160 mmHg) hypertension increase the risk of AD by 18% and 25%, respectively(Lennon et al., 2019).However, the exact mechanisms by which hypertension may contribute to AD are still being investigated.Hypertension may damage cerebral vasculature endothelium, resulting in altered cerebral autoregulatory mechanisms that lead to cerebral hypoperfusion and, subsequently, cognitive deficits (Hannesdottir et al., 2009).One of the mechanisms causally linking high blood pressure with disrupted BBB involves hypertension-induced oxidative stress in cerebral vessels.This leads to increased activity of matrix metalloproteinases, which degrade tight junction proteins of the BBB(Toth et al., 2015).When released by glial cells, these enzymes can also damage myelin and synapses (Szklarczyk and Conant, 2010).Furthermore,cerebrovascular injury induced by hypertension has been shown to impair the integrity of vascular BBB in animal models, leading to increased deposition of amyloid-β (Kruyer et al., 2015; Shih et al., 2018).Hypertension has also been shown to be associated with inflammation (Xiao and Harrison, 2020), which may represent another mechanism by which this modifiable factor increases the risk of developing AD.
Hypertension and Inflammation
Increasing evidence suggests that vascular and inflammatory responses under hypertensive conditions are closely related (Pauletto and Rattazzi,2006; Sprague and Khalil, 2009).Vascular injury upregulates expression of adhesion molecules by endothelial cells, facilitating the recruitment of inflammatory cells, including monocytes, macrophages, and lymphocytes,that secrete a range of pro-inflammatory mediators, such as TNF, interferon-γ,interleukins (IL)-1, -2 and -6, and monocyte chemoattractant protein-1 (MCP-1/CCL2) (Sprague and Khalil, 2009).A disrupted BBB allows extravasation of plasma proteins, such as fibrinogen, thrombin, and immunoglobulins,which activate glial cells directly.Reactive glia release neurotoxins, hindering synaptic transmission and causing damage to neurons.An alternative mechanism facilitating neuroinflammation is the impaired clearance function of the glymphatic system caused by elevated microvascular pressure, which promotes the development of cerebral microhemorrhages.Ungvari et al.(2021) postulate that exacerbation of neuroinflammation is one of the key factors causally linked to the development of AD in hypertensive elderly individuals, as well as to cognitive impairment that accompanies aging.This conclusion is based mainly on preclinical data, such as a study by Sadekova et al.(2018) who demonstratein vivothat the hemodynamic changes induced by arterial stiffness cause oxidative stress-dependent activation of microglia and astrocytes in the hippocampus of mice.
Conversely, inflammatory changes in endothelial cells are triggered by hypertension-induced macrophage-derived exosomes (Osada-Oka et al.,2017).Epidemiological studies support these observations by demonstrating higher plasma levels of TNF, IL-6, CCL2, and the inflammatory marker C-reactive protein in patients with pre-hypertension, as well as hypertension without cardiovascular disease, compared to normotensive individuals(Pauletto and Rattazzi, 2006).
Similar inflammatory mechanisms link hypertension with atherosclerosis.The inflammatory response associated with atherosclerosis increases permeability of vasculature, allowing for the accumulation of T-cells and monocytes/macrophages within the intimal layer of blood vessels.These inflammatory cells then release pro-inflammatory molecules that further propagate vascular inflammation (reviewed by Tabas and Lichtman, 2017).A recent study comparing monocytes isolated from hypertensive and normotensive subjects, using next-generation RNA sequencing approach, identifies 60 genes with differential expression.Many of these genes are associated with the IL-1β signaling, indicating overstimulated inflammasome pathway in peripheral blood cells during hypertension (Alexander et al., 2019).The nucleotide-binding domain, leucine-rich-containing family pyrin domain containing 3 (NLRP3) inflammasome, which is engaged in the activation of caspase-1 and the maturation of IL-18 and IL-1β, has been implicated in inflammatory processes associated with hypertension (De Miguel et al.,2021).Thus, patients with essential hypertension exhibit increased serum levels of IL-1β, which are associated with increased risk for developing atherosclerosis (Dalekos et al., 1997).IL-1β has also been shown to causally link atherosclerotic processes with CNS inflammation.For example, Denes et al.(2012) demonstrate that diet-induced atherosclerosis in mice is associated with severe cerebrovascular inflammation, including microglial activation,which is significantly reduced in IL-1 type 1 receptor-deficient animals and by systemic neutralization of IL-1β using antibodies.
Another mechanism linking hypertension, neuroinflammation and AD involves the nicotinamide adenine dinucleotide (NAD+)-mitophagy axis.NAD+is a co-factor for several key metabolic pathways and it is also an inducer of mitophagy, a process that is essential for the removal of defective mitochondria.Aging is associated with reduced NAD+levels, which can lead to reduced clearance of damaged mitochondria disrupting biochemical homeostasis of cells.Age-related dysfunction of mitophagy contributes to the development of neurological, as well as cardiovascular, diseases,including endothelial damage, atherosclerosis, and hypertension (Das et al.,2018; Fang and Tao, 2020; Morciano et al., 2020).Accumulation of damaged mitochondria and the associated oxidative stress have been postulated as one of the key mechanisms linking aging with an increased risk of AD (Grimm et al., 2016).In a recent study, Fang et al.(2019) demonstrate decreased mitophagy in microglia from the hippocampus of APP/PS1 AD model mice,which is associated with their pro-inflammatory activation.Stimulants of mitophagy reduce microglial reactivity assessed by measuring their TNF secretion.Interestingly,in vivoadministration of mitophagy inducers reduces NLRP3 inflammasome expression and activation that is upregulated in corticaltissues of AD mice, lowers levels of brain IL-1β, IL-6, and TNF, and ameliorates amyloid-β pathology and cognitive decline in these animals.Therefore,NAD+-mitophagy defects could be a common mechanism contributing to hypertension-related AD and neuroinflammation.
Increased NLRP3, IL-1β, and oxidative stress have been demonstrated in the hypothalamic paraventricular nucleus (PVN) of the Dahl salt-sensitive rats receiving a high-salt diet compared to rats fed normal diets (Qi et al., 2016).The hypothalamus is essential for blood pressure regulation and is significantly affected by persistent hypertension.In salt-induced pre-hypertensive rats, upregulation of NLRP3 and increased inflammatory mediators CCL2,CXC chemokine receptor type 3, and vascular cell adhesion molecule 1 in the PVN are accompanied by elevated numbers of microglia and T-cells,demonstrating a possible link between hypertension and neuroinflammation(Wang et al., 2018).Furthermore, an increase in BBB permeability may occur in hypertension, as well as normal aging, and may allow for the more widespread influx of peripheral immune cells and pro-inflammatory mediators into the brain parenchyma where they could contribute to the neuroinflammation seen in AD (Senatorov et al., 2019; Xiao and Harrison, 2020).
Hypertension and Neuroinflammation in Alzheimer’s Disease Models
Numerous animal studies have been performed to investigate whether inflammation may mediate the pathological consequences of hypertension in AD (Table 1).Kruyer et al.(2015) used Nω-nitro-L-arginine methyl ester to chemically induce chronic hypertension in the Tg-SwDI mouse model ofAD, demonstrating that high blood pressure accelerates BBB leakage and cognitive deficits, and induces cerebrovascular microgliosis 6 months after treatment.Increased microvascular deposition of amyloid-β and hippocampal neurodegeneration are also observed in these mice.Carnevale et al.(2012)used hypertensive mice with transverse aortic coarctation to demonstrate microglial activation and IL-1β upregulation occur as early as three weeks after hypertensive insult, followed by increased amyloid-β deposition at four weeks.This observation indicates CNS inflammation as the causal link between hypertension-induced alterations in the cerebral microvasculature and the pathogenetic mechanism of AD.Raz et al.(2019) reported reduced cerebral blood flow, decreased cortical oxygen levels, and reduced oxygen carrying neuronal neuroglobin in the spontaneously hypertensive and stroke-prone rat fed a high-salt, low-protein Japanese permissive diet, which is suggestive of chronic cerebral hypoperfusion.In addition, they observe a corresponding increase in active IL-1β, reactive oxygen species, hyperphosphorylated tau protein, and neurodegeneration in the hippocampus of these animals.The authors suggest that hypertension causes damage to the cerebral microvasculature resulting in cerebral hypoxia and hypoperfusion, which in turn triggers a neuropathological cycle of microglial activation, production of reactive oxygen species, and BBB dysfunction.Shih et al.(2018) examine whether hypertension accelerates the onset of AD using two different animal models.First, hypertension is induced in Lanyu miniature pigs by performing an abdominal aortic constriction surgery, followed by assessment of AD-related pathologies at one, two, and three months after the operation.Hypertension can be detected in the pigs starting one month after the surgery, and the hippocampal levels of phosphorylated tau and amyloid-β are increased after three months.The second model involves using the “two-kidney-one-clip”procedure to induce hypertension in 3xTg AD mice.Hypertension in these animals can be detected one month following the operation; it is accompanied by increased hippocampal levels of amyloid-β and phosphorylated tau, BBB leakage, activation of microglia, measured as increased number of cells expressing ionized calcium binding adaptor molecule 1 (Iba-1), and impairment in hippocampus-dependent learning and memory (Shih et al., 2018).These results indicate that hypertension accelerates the onset of AD pathology and that CNS inflammation may be one of the possible mechanistic links.
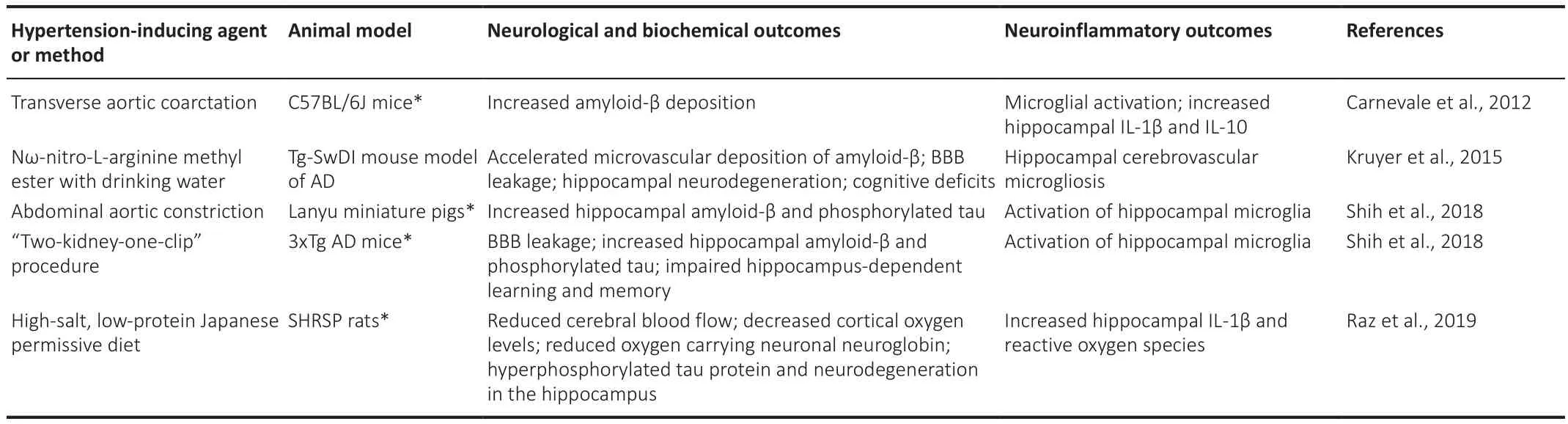
Table 1 |Neuroinflammatory processes in animal models of hypertension
Angiotensin II Regulates Blood Pressure and Glial Activation
Angiotensin (Ang)-II is an octapeptide hormone of the renin-angiotensin system (RAS) involved in regulating blood pressure.Renin is released when a drop in blood pressure is detected by the kidneys and leads to the production of Ang-II, which increases blood pressure and blood volume by causing vasoconstriction and promoting salt-retention by the kidneys.As such, Ang-II has been clinically associated with the development of hypertension.Ang-II also connects the RAS and neuronal control of blood pressure by binding to Ang-II type 1 receptors (AT1R) found in the CNS.Persistent activation of AT1R of the circumventricular organs and the cardioregulatory nuclei stimulates vasopressin secretion, increases sympathetic nervous system outflow, and reduces baroreflex sensitivity (Mowry and Biancardi, 2019).In addition,stimulation of the PVN by Ang-II causes excitatory effects on the renal sympathetic nervous system, which induces renin release (Ferguson et al.,2008).
While the “classical” RAS functions as a circulating humoral system, locally acting RAS systems have been described for several tissues, including the brain where different CNS cell types not only express the two main ATRs but can also locally produce Ang-II.There is also interplay between the systemic RAS and CNS cells (reviewed by Labandeira-Garcia et al., 2017).Notably, subcutaneous infusion of Ang-II for four weeks leads to microglial activation in the PVN,assessed by their shortened processes and upregulated CD11b expression,which is accompanied by increased TNF, IL-1β, and IL-6 mRNA levels (Shi et at., 2010).The key contribution of TNF to hypertension is demonstrated by Sriramula et al.(2008), who show that subcutaneous administration of Ang-II for two weeks increases blood pressure in wild-type mice but has no effect in TNF knockout mice.In addition, the hypertensive effects of Ang-II are induced in TNF knockout mice after treatment with recombinant TNF.A recent study by Iulita et al.(2018) demonstrates that systemic administration of Ang-II to mice increases hippocampal gliosis measured as elevated levels of Iba-1 and glial fibrillary acidic protein, which are markers of microgliosis and astrogliosis, respectively.Phenylephrine, an agent that elevates blood pressure independently of the RAS system, increases hippocampal glial fibrillary acidic protein levels but has no effect on Iba-1.These animal studies provide evidence that inflammatory cytokines are essential for the blood pressure regulation by RAS, but also demonstrate that hypertension can lead to neuroinflammation contributing to the development of AD.Furthermore, these findings indicate that Ang-II may act as a pro-inflammatory mediator and represent the link between hypertension and neuroinflammation.Accordingly, control of Ang-II levels and reducing microglial activation may represent strategies to reduce neuroinflammation associated with hypertension.
Controlling Neuroinflammation Associated with Hypertension
Accumulating evidence from controlled clinical trials indicates that antihypertensive treatment delays cognitive decline and reduces risk of dementia.While lowering blood pressure is likely the main beneficial effect of such therapies, experimental data reveal additional neuroprotective and antidegenerative effects for some classes of antihypertensives, such as calcium channel blockers and drugs acting on the RAS (reviewed by Desideri and Bocale, 2020).Furthermore, animal studies demonstrate that antihypertensive drugs can be used to control neuroinflammation (Table 2).For example, the smooth muscle relaxant hydralazine not only lowers blood pressure, but also inhibits the associated hippocampal TNF production as well as reduces Iba-1 and glial fibrillary acidic protein levels in Ang-II-induced hypertensive mice (Iulita et al., 2018).Microglia have been shown to express ACE as well as AT1R and AT2R, and are considered part of the RAS of the brain (Zhang et al., 2016; Labandeira-Garcia et al., 2017).An ACE inhibitor captopril, which is used as an antihypertensive drug, has been shown to inhibit secretion of nitric oxide and TNF by bacterial lipopolysaccharide-stimulated rat glial cell cultures.Intranasal administration of this drug to 5xFAD AD model mice for 3.5 weeks results in significant reduction of cortical CD11b staining, a marker of activated microglia, compared to saline-treated mice.Interestingly, after two months of captopril treatment, these mice display reduction in both CD11b and amyloid-β in cortical areas.However, after seven months, while there is further reduction in amyloid-β staining, the levels of CD11b are no longer different from saline-treated 5xFAD mice cortices (Asraf et al., 2018).The proinflammatory effects of Ang-II in the PVN are mediated by AT1R, which is the predominant form expressed in this brain region; meanwhile, agonists of AT2R have been demonstrated to possess anti-inflammatory effects.Cell culture systems and animal models have been used to demonstrate that AT2R agonists,such as C21 and CGP42112, and the AT1R antagonist losartan downregulate microglial activation and inflammatory mediators IL-1β, IL-6, CCL2, CCL3,CXCL10 and TNF (Stegbauer et al., 2009; Elsaafien et al., 2020).It has been proposed that activation of microglial AT1R, which are upregulated under CNS inflammatory states, induces their polarization towards the pro-inflammatory and cytotoxic M1 phenotype, while activation of AT2R shifts microglia towards the M2 inflammation resolving phenotype; therefore, both AT1R antagonists and AT2R agonists have been used to reduce neuroinflammation in AD models(reviewed by Labandeira-Garcia et al., 2017).
Another pharmacological agent successfully used to control neuroinflammation and hypertension is the antibiotic minocycline, which is also known for its anti-inflammatory and neuroprotective properties.Intracerebroventricular infusion of minocycline prevents hypertension induced by subcutaneous administration of Ang-II.Furthermore, minocycline reduces mRNAs for TNF, IL-1β, IL-6 in the PVN and decreases the number of activated microglia in this brain region of Ang-II-treated animals (Shi et al., 2010).Table 2 lists several classes of drugs that can be used to control neuroinflammation associated with hypertension.Combining these differentially acting pharmacological agents may lead to synergistic beneficial effects; however, such combination therapies will require careful preclinical and clinical studies since, for example, adverse interactions between non-steroidal anti-inflammatory drugs and several different classes of antihypertensive medications have been documented (Fournier et al., 2014).
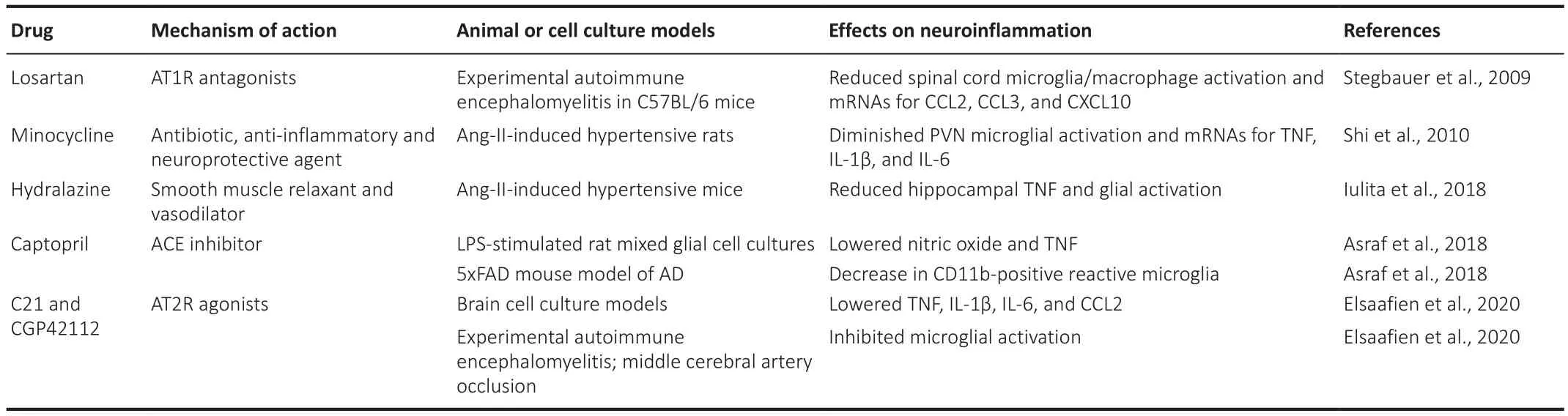
Table 2 |Effects of antihypertensive drugs in neuroinflammatory models in vitro and in vivo
Conclusions and Future Directions
Current evidence implicating neuroinflammation as the mechanistic link between midlife hypertension and increased risk of AD is almost exclusively based on cell culture studies and animal models of both these pathologies(Figure 1).Direct proof of this concept will require clinical studies due to the inadequacy of current AD models, which is increasingly recognized as one of the main reasons for failure to translate highly effective treatments achieved in animal models to human AD (Drummond and Wisniewski, 2017).The majority of clinical trials in AD (more than 1000) have studied therapeutic agents and strategies aimed at amyloid-β, tau protein or dysregulated neurotransmitters (reviewed by Liu et al., 2019).To the best of our knowledge only a single clinical trial testing the antihypertensive ACE inhibitor, ramipril,in subjects at risk for AD has been published (Wharton et al., 2012).This pilot study involving 14 cognitively healthy individuals with mild, or stage I hypertension, demonstrates four months of ramipril therapy significantly improves blood pressure and inhibits ACE activity in cerebrospinal fluid;however, ramipril has no effect on cognitive functions or cerebrospinal fluid levels of amyloid-β and tau.Larger clinical trials over longer treatment periods are highly warranted to test the hypothesis that antihypertensive therapies are beneficial in AD and that such intervention ameliorates neuroinflammation.
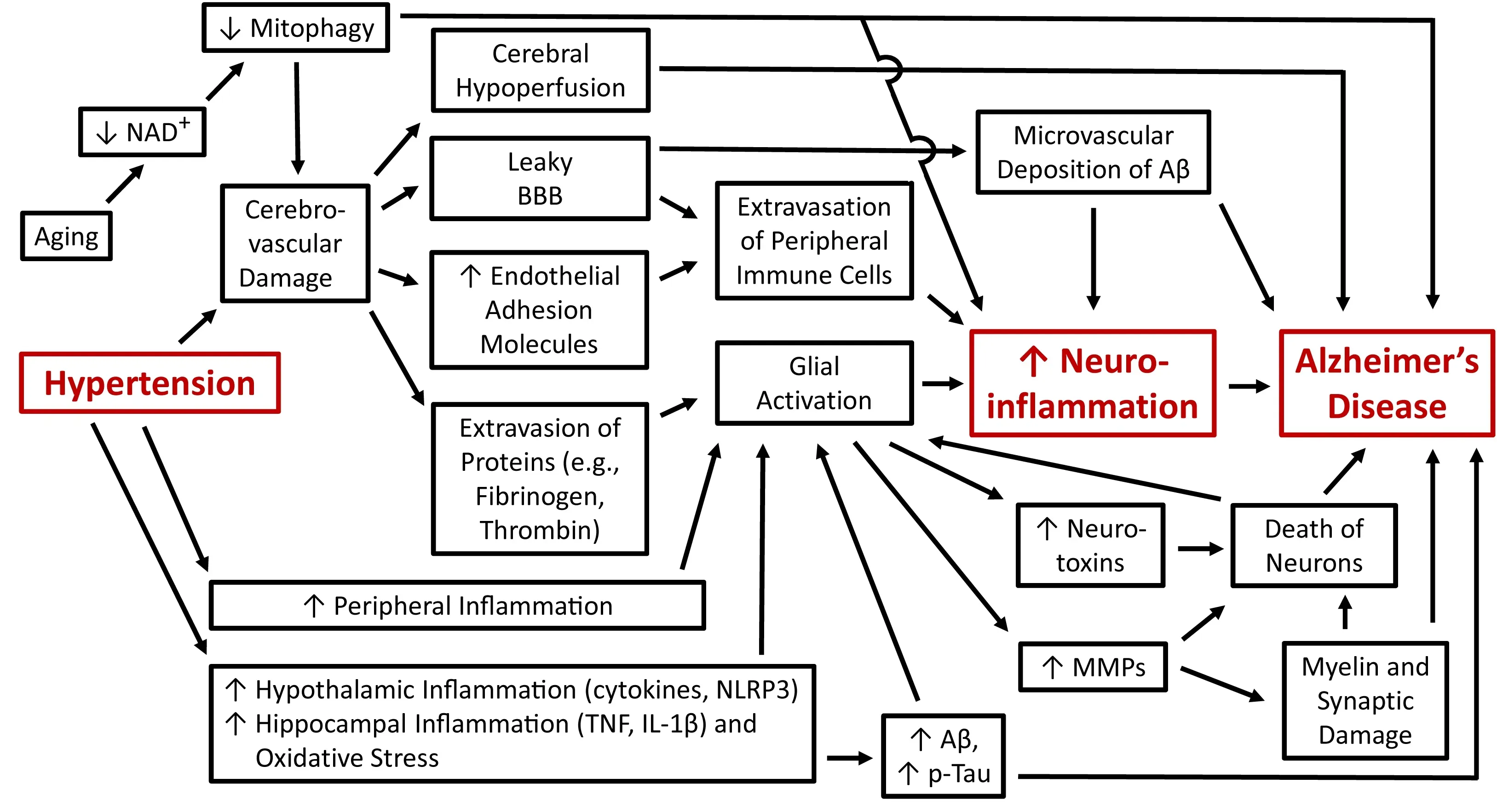
Figure 1| A schematic diagram showing the cellular and molecular mechanisms discussed in this article that link hypertension, neuroinflammation and Alzheimer’s disease.
Further pre-clinical and clinical studies are also needed to conclusively establish the causal contribution of neuroinflammatory processes to the observed link between hypertension and AD since most of the animal studies and all human research performed to date only demonstrate correlations between hypertension, neuroinflammatory parameters and AD symptoms.Only select pre-clinical research has employed the pharmacological approach (Sadekova et al., 2018), longitudinal studies (Carnevale et al.,2012), and transgenic animals (Denes et al., 2012) to demonstrate that neuroinflammatory mechanism are not just passive bystanders, but active contributors to hypertension-induced cognitive decline and AD pathogenesis.
The main challenge for such clinical studies is visualization of neuroinflammation in human subjects.In this regard, the recent study by Low et al.(2020), who use [11C]PK11195 positron emission tomography (PET) to visualize microglial activation in healthy controls and patients with mild AD or amyloid-β-positive mild cognitive impairment, is notable.Their PET imaging demonstrates that microglial activation is associated with cerebral small vessel disease assessed by MRI, particularly with the small vessel disease subtype of hypertensive arteriopathy.Since hypertension and BBB leakage have been implicated in etiology of hypertensive subtype of small vessel disease, this study may provide clinical evidence of neuroinflammation resulting from elevated blood pressure even though, as pointed out by the authors of this study, their data do not imply causality and only establish association (Low et al., 2020).
Future studies investigating neuroinflammation in hypertensive patients could also employ specific PET tracers to visualize astrogliosis.The best characterized reactive astrocyte-specific radiotracer is [11C]-deuterium-L-deprenyl ([11C]DED), which binds to astrocyte monoamine oxidase-B (Carter et al., 2019).Future multi-tracer PET studies can be used to not only visualize microglia and astrocyte activation but to consider it in the context of developing AD pathology in at risk subjects by monitoring changes in cerebral glucose metabolism, brain neurotransmitter systems (cholinergic, dopaminergic and serotonergic), and both amyloid-β and tau aggregation (Nordberg et al., 2010).Even though a routine use of such studies for clinical diagnosis and AD risk assessment is unlikely, they can be used in research settings to conclusively establish the link between hypertension, neuroinflammation and AD pathogenesis, ascertain the causal relationships between these pathophysiological mechanisms, as well as monitor any potential disease modifying effects of anti-inflammatory and antihypertensive medications.Notably, in a very small preliminary study involving only eight subjects, Drake et al.(2011) used [11C]PK11195 PET to demonstrate that subjects, presenting with several (3-4) risk factors for stroke (including hypertension), display increased microglial activation compared to patients with just 1-2 risk factors.Finally, due to the recent significant success with the developing human pluripotent stem cell-derived microglia and astrocyte cultures, as well as brain organoid systems containing all main CNS cell types (reviewed by Sidhaye and Knoblich, 2021), human-specific tissue culture systems can be set up in the search for effective inhibitors of neuroinflammation, which could be used to prevent development of cognitive impairment and AD in at risk subjects with history of hypertensive disease.
Acknowledgments:The authors would like to thank all members of the Laboratory of Cellular and Molecular Pharmacology, UBC Okanagan campus,Canada for helpful discussions and comments on the manuscript.
Author contributions:EB wrote the original draft; AK revised and expanded the manuscript; EB and AK contributed to editorial changes in the manuscript,read and approved the final version of the manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Availability of data and materials:All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Renato Bernardini, University of Catania, Italy; Evandro Fei Fang, University of Oslo and Akershus University Hospital, Norway.
Additional file:Open peer review reports 1 and 2.
- 中国神经再生研究(英文版)的其它文章
- Interplay of SOX transcription factors and microRNAs in the brain under physiological and pathological conditions
- Cerebellar pathology in motor neuron disease:neuroplasticity and neurodegeneration
- An atypical ubiquitin ligase at the heart of neural development and programmed axon degeneration
- The endogenous progenitor response following traumatic brain injury: a target for cell therapy paradigms
- The relationship between amyloid-beta and brain capillary endothelial cells in Alzheimer’s disease
- Telomerase and neurons: an unusual relationship