
猴欢喜叶绿体全基因组及杜英科系统地位分析
2022-03-17 01:05王一麾谢宜飞张志翔金佳怡邱相东童阳
广西植物 2022年1期
王一麾 谢宜飞 张志翔 金佳怡 邱相东 童阳




摘 要: 最新的分子系統发育(APG IV)研究中以猴欢喜属(Sloanea L.)为代表的杜英科(Elaeocarpaceae)所在的酢浆草目(Oxalidales)被置于豆类分支(Fabids),且与卫矛目(Celastrales)、金虎尾目(Malpighiales)组成一支(COM分支),但支持率较低。为提高COM分支支持率,该文以杜英科猴欢喜属猴欢喜(Sloanea sinensis)为材料,应用Illumina Hiseq 2500对猴欢喜叶绿体基因组进行测序,并通过Geneious 11.0,PGA和Shiny软件进行组装,注释和绘制基因组图谱,之后使用MISA和IRscope软件对叶绿体基因组特征进行分析,并使用PhyloSuite软件构建豆类分支系统发育树。结果表明:(1)猴欢喜叶绿体基因组全长157 546 bp,GC含量为37.0%,包含一对25 984 bp的反向重复区、大单拷贝区(87 903 bp)和小单拷贝区(17 675 bp)。(2)在猴欢喜叶绿体基因组中有113个非重复基因,包括79个蛋白质编码基因、4个rRNA基因和30个tRNA基因;共检测到81个SSR位点,其中大部分是单核苷酸重复序列;通过IR边界区比较分析,发现日本杜英(Elaeocarpus japonicus)和猴欢喜在LSC/IRb和IRa/LSC边界上存在明显差异。(3)构建豆类分支叶绿体全基因组最大似然法系统发育树,猴欢喜同日本杜英亲缘关系最近,与酢浆草科(Oxaliadaceae)阳桃(Averrhoa carambola)、红花酢浆草(Oxalis corymbosa)和O. drummondii聚为一支,支持杜英科归于酢浆草目并且处在豆类分支中,且卫矛目、酢浆草目和金虎尾目演化支(COM分支)支持率高达100%。该研究基于猴欢喜叶绿体基因组进一步确定了杜英科和COM分支的系统地位。
关键词: 猴欢喜, 叶绿体基因组, 系统发育分析, 基因组比较, COM分支
中图分类号: Q941
文献标识码: A
文章编号: 1000-3142(2022)01-0039-10
收稿日期: 2021-04-23
基金项目: 国家自然科学基金(31110103911, J1310002)[Supported by the National Natural Science Foundation of China(31110103911, J1310002)]。
第一作者: 王一麾(1997-),硕士研究生,主要从事被子植物系统发育学研究,(E-mail)535401842@qq.com。
*通信作者: 谢宜飞,博士,讲师,主要从事木本被子植物系统发育学研究,(E-mail)xiey-f@foxmail.com。
The complete chloroplast genome of Sloanea sinensis
and the systematic status of Elaeocarpaceae
WANG Yihui1, XIE Yifei1,2*, ZHANG Zhixiang3, JIN Jiayi1, QIU Xiangdong1, TONG Yang1
( 1. School of Life Sciences, Gannan Normal University, Ganzhou 341000, Jiangxi, China; 2. Nanling Herbarium, Gannan Normal University,
Ganzhou 341000, Jiangxi, China; 3. School of Ecology and Nature Conservation, Beijing Forestry University, Beijing 100083, China )
Abstract: Angiosperm Phylogeny Group IV (APG IV) revealed Elaeocarpaceae represented by Sloanea sinensis belonging to Oxalidales includes the COM clade with Celastrales and Malpighiales, while the status of COM clade is not well supported based on phylogenetic calculation results of multi-molecular fragments. In order to improve the bootstrap of COM clade, with Sloanea sinensis (genus Sloanea, family Elaeocarpaceae) as materials, we used Illumina Hiseq 2 500 platform to sequence, and then assembled, annotated, and analyzed by Geneious 11.0 PGA and Shiny. Analysis of chloroplast genome characteristics was used by MISA and IRscope. The phylogenetic tree of Fabids was reconstructed by using PhyloSuite. The results were as follows: (1) The complete chloroplast genome of Sloanea sinensis was 157 546 bp in length including two inverted repeats (IRs) of 25 984 bp, which were separated by large single copy (LSC) and short single copy (SSC) of 87 904 bp and 17 674 bp, respectively. The GC content was 37.0%. (2) The genome encoded 113 functional genes, including 79 protein-coding genes, 30 tRNA genes, and 4 rRNA genes. 81 SSR loci were detected in the S. sinensis genome, and most of SSR was composed of nucleobase A and T. Through IR expansion and contraction analysis, we found that there were obvious differences between Elaeocarpus japonicus and Sloanea sinensis in LSC/IRB and IRA/LSC boundaries. (3) The whole chloroplast genome phylogenetic studies showed Sloanea sinensis and Elaeocarpus japonicus were sister to Averrhoa carambola, Oxalis corymbosa and O. drummondii. Sloanea sinensis and Elaeocarpus japonicus representing Elaeocarpaceae belonged to Oxalidales, Fabids, and a strongly support for COM clade in Fabids group. Based on the chloroplast genome of genus Sloanea, the phylogenetic status of Elaeocarpaceae and the COM clade is confirmed.
Key words: Sloanea sinensis, chloroplast genome, phylogenetic analysis, genome comparison, COM clade
叶绿体是色素、脂类物质、激素和核糖体等合成的重要细胞器(Palmer 1985; Shinozaki et al., 1986)。叶绿体的遗传物质为叶绿体基因组,通常表现为四联体结构,由两个倒位形成的高度保守的重复区域(IRa和IRb),一个大单拷贝区域和另一个小单拷贝区域(分别为LSC和SSC)组成(Pogson et al., 1847; Raman & Park, 2015; Cheng et al., 2017)。叶绿体基因组因其结构保守,同核基因更易于获得且叶绿体基因组一般是单亲遗传,易于分析(陶晓丽等,2017;Li et al., 2018;Zhang et al., 2018;Jeon & Kim, 2019)。随着NCBI数据库中叶绿体基因组数据的日益增长,为叶绿体比较基因组学提供了丰富的材料,特别是系统发育分析、分子系统地理学、单倍型分析和转基因遗传改良方面应用潜力极大(Zhang et al., 2017)。杨亚蒙等(2019)基于34个葡萄属(Vitis)叶绿体全基因组数据重建系统发育树,结果与传统分类一致;Bhati等(2018)应用Illumina Miseq平台对茴香(Foeniculum vulgare)叶绿体基因组进行测序比较,共获得23对分子标记引物,其中21对引物是伞形科植物的有效微卫星;Marechal & Brisson(2010)研究发现IR边界区的扩张与收缩是叶绿体基因组结构的重要特征;Su等(2020)比较小麦属的叶绿体基因组结构中发现IR区较其他物种的遗传差异大,变异明显,这可能与二粒系小麦的遗传多样性相关。
以形态学特征为主的解剖学证据曾将杜英科归属于锦葵目、红树目等(Takhtajan, 1980;张宏达, 1989;Thorne, 2000)。Crayn等(2006)基于分子数据推断杜英科(Elaeocarpaceae)与瓶子草科(Cephalotaceae)、槽柱花科(Brunelliaceae)的亲缘关系最近,应隶属于豆类酢浆草目。最新的APG IV系统把包括杜英科的酢浆草目、卫矛目、金虎尾目(COM分支)放置于豆类中,但COM分支的支持率较低(Chase et al., 2016)。
近年来有学者开展了杜英科属间的系统发育关系研究,利用rbcL片段和trnL-trnF间隔区构建了杜英科Aceratium、Elaeocarpus、Plotytheca、Crinodendron、Valea和Sloanea 的属间系统发育关系,划分为(Crinodendron + Valea)+ Sloanea 分支和(Aceratium + Elaeocarpus)+ Plotytheca分支(Bradford & Barnes, 2011)。根据形态和分子证据,将杜英科分为Sloanea alliance (Vallea, Aristotelia and Sloanea)、Tremandraceous genera (Platytheca, Tetratheca and Tremandra)和Elaeocarpus alliance (Sericolea, Aceratium and Elaeocarpus)三个分支(Phoon, 2015;谢宜飞, 2018)。Sloanea alliance (Vallea, Aristotelia and Sloanea) 分支主要分布在南美洲西部和北部、东南亚和澳洲东部。本研究以猴欢喜为材料,通过叶绿体基因组高通量测序、组装和基因注释,利用猴欢喜和日本杜英叶绿体全基因组系统发育树进一步确定杜英科和COM分支的位置。
1 材料与方法
1.1 實验材料
测序材料为猴欢喜(Sloanea sinensis),于2017年4月22日在云南省麻栗坡县天保乡(104°43′12″ E、23°0′36″ N)采摘其新鲜嫩叶。材料凭证标本保存于北京林业大学标本馆(BJFC),采集号为谢宜飞、王磊XW1956,鉴定人为谢宜飞。
1.2 基因组DNA提取及测序
采用改良的十六烷基三甲基溴化铵(CTAB)法提取猴欢喜总DNA(Li et al., 2009),之后利用琼脂糖凝胶电泳和NanoDrop-2000微量分光光度计检测DNA纯度和浓度并将猴欢喜的DNA送至北京睿博兴科生物技术有限公司,并使用Illumina HiSeq (TM) 2000进行高通量测序。将得到的原始图像数据文件经CASAVA碱基识别(base calling)分析转化为原始测序序列(raw data),将原始测序序列过滤掉带接头的、低质量的reads,获得6.04 Gb的clean reads,并且命名为DMS14627-S.fq.gz文件(Cock et al., 2010; Hansen et al., 2010)。
1.3 基因组组装与注释
经Geneious 11.0软件Trim Ends去除接头两端低质量序列后的高质量分析序列,以Brunellia trianae (槽柱花科槽柱花属,GenBank登录号:MN585217)叶绿体基因组为参考序列进行组装和注释。利用SPAdes v3.6.1在默认参数下进行从头拼接,并生成一系列Contigs (Prjibelski et al., 2020),将长度大于1 000 bp的Contigs用于叶绿体基因组组装,匹配连接构建完整的叶绿体基因组序列(Kearse et al., 2012),二代测序技术补齐组装后的空缺。使用PGA基因组注释工具对猴欢喜叶绿体基因组进行功能注释,并使用Shiny软件绘制猴欢喜叶绿体基因组图谱(Liu et al., 2018; Zheng et al., 2020)。测序后的clean reads上传至NCBI的SRA数据库(PRJNA661695, SRR12599358),使用Bankit将注释好的序列提交至NCBI,获得Genbank登录号:MW004670。
1.4 叶绿体基因组特征分析
猴欢喜叶绿体基因组序列为材料,使用MISA软件(http://pgrc.ipk-gatersleben.de/misa/misa.ht-ml)鉴定猴欢喜叶绿体基因组中简单重复序列(SSR),参数采用默认值,即单核苷酸、二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸的最小重复次数分别为 10、6、5、5、5、5,2 个 SSR之间的最小距离为 100 bp (Sahu et al., 2012)。使用IRscope (https: ∥irscope.shinyapps.io/irapp/) 绘制猴欢喜、阳桃、Oxalis corniculate、O. drummondii和日本杜英叶绿体基因组边界(Amiryousefi et al., 2018)。
1.5 序列比对和系统发育分析
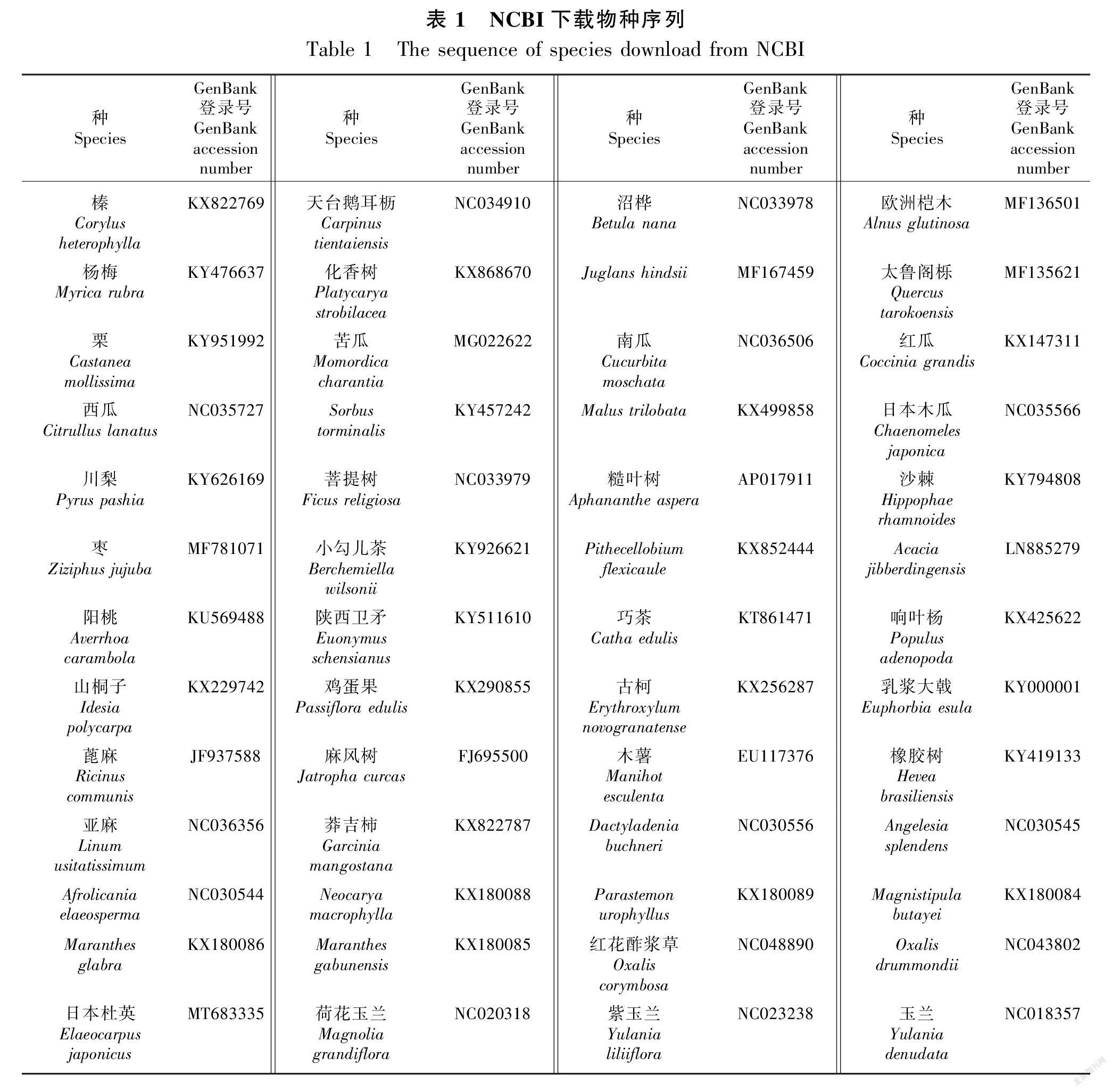
从NCBI数据库下载榛(Corylus heterophylla)、天台鹅耳枥(Carpinus tientaiensis)、沼桦(Betula nana)等52个物种完整叶绿体基因组序列。其中,9个物种来自壳斗目,4个物种来自葫芦目,9个物种来自蔷薇目,2个物种来自豆目,4个物种来自酢浆草目,2个物种来自卫矛目,19个物种来自金虎尾目,3个物种来自木兰目(表1)。利用MEGA 7.0软件比对序列,去除两端不整齐序列(Kumar et al., 2016)。使用PhyloSuite软件ModelFinder进行模型分析,并构建最大似然法系统发育树。按照自展值bootstrap为1 000,运行最长时间为168 h,其他参数为默认值进行计算。Tree文件使用软件FigTree v1.3读取(Price et al., 2009; Zhang et al., 2020)。
2 结果与分析
2.1 基因组基本特征
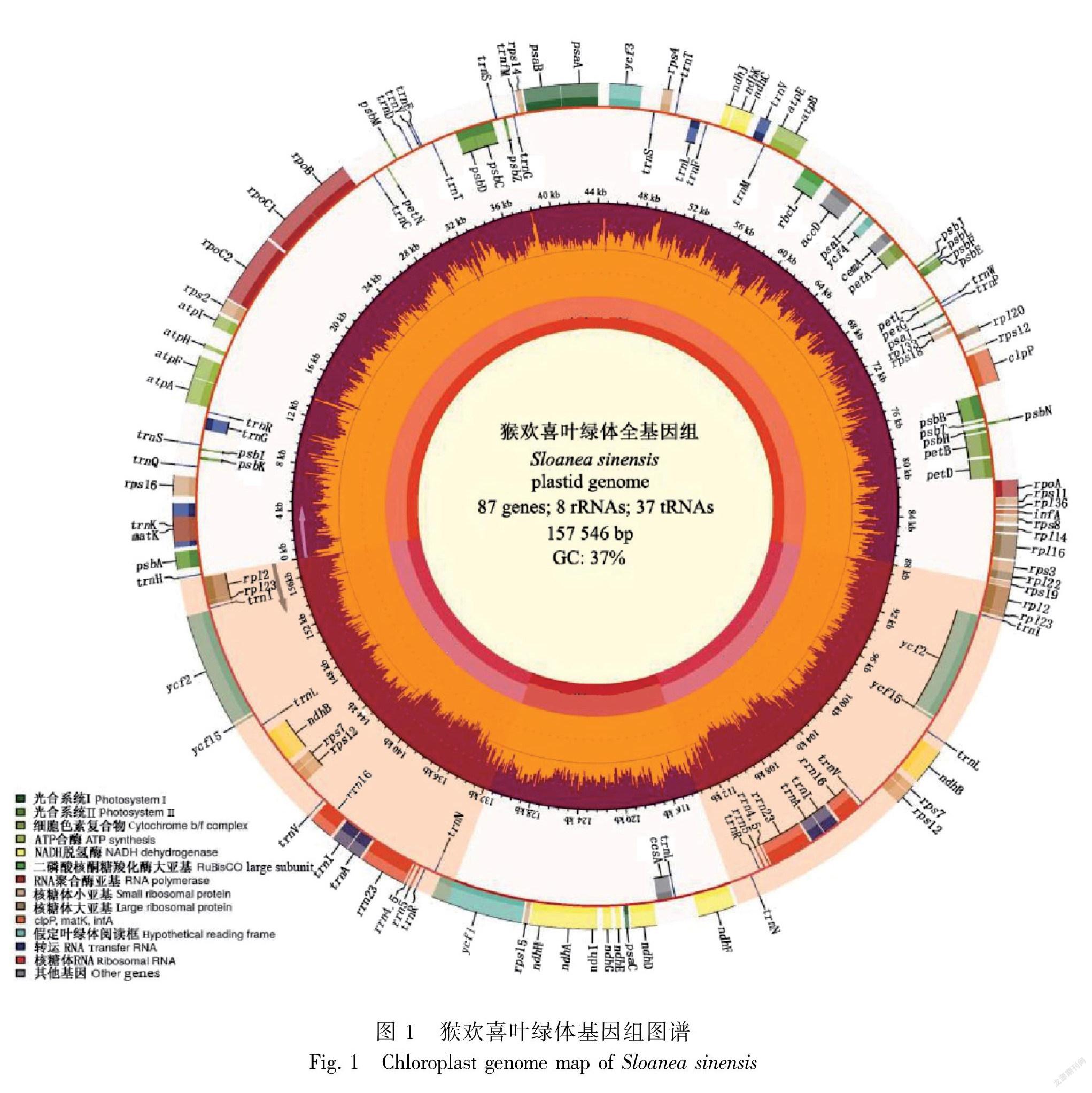
猴欢喜叶绿体基因组全长157 546 bp, LSC、SSC与IR的长度分别为87 903 bp、17 675 bp、25 984 bp。全基因组的GC含量为37.0%,其中GC含量最高的是反向重复区(42.9%),大单拷贝区和小单拷贝区分别为35.1%和31.4%(表2,图1)。猴欢喜叶绿体基因组共包括132个基因,非重复基因113个(表3)。
2.2 SSR分析
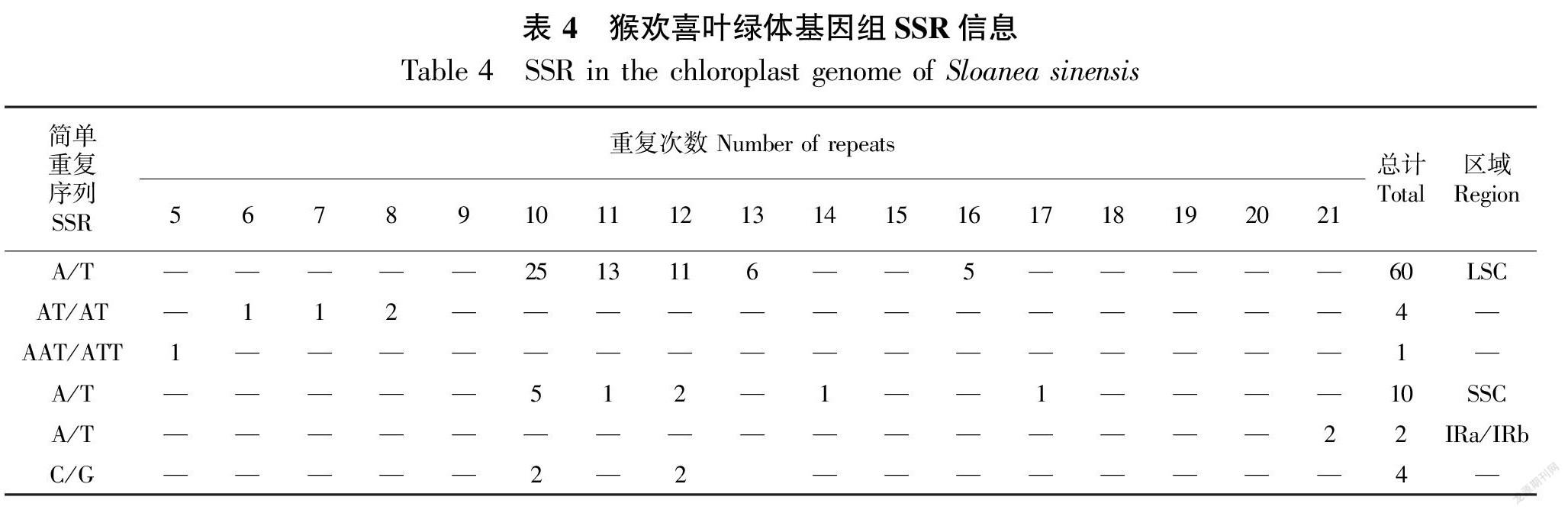
本研究发现猴欢喜叶绿体基因组中存在81个SSR(Simple Sequence Repeat,简单重复序列标记),其中大部分是单核苷酸重复序列(76个,93.83%),二核苷酸重复序列有4个(4.94%),三核苷酸重复序列只有1个(1.23%),未检测到四核苷酸、五核苷酸和六核苷酸序列。88.89%的SSR由A或T组成,这表明SSR基因组偏向A/T基因。SSR以长度10~12 bp的短序列为主,占全部的75.31%,16 bp以上的仅占9.88%(表4)。LSC区域内核苷酸重复序列达65个(80.25%),其中大部分是单核苷酸重复序列。SSC区域内核苷酸重复序列有10个(12.35%),且都是单核苷酸重复序列,而两个IR区内有6个(7.41%)单核苷酸重复序列。
2.3 IR边界区分析
借助IRscope工具生成5种酢浆草目植物的边界图(图2),5种植物IRa/SSC边界的基因分布相似。其中,猴欢喜和日本杜英的ndhF基因距离IRb/SSC边界较远。在LSC/IRb和IRa/LSC边界上,猴欢喜同3种酢浆草属植物所含基因相同(rps19和rpl2),而日本杜英在这两个边界上是rps3和rpl22。
2.4 系统发育分析
将猴欢喜、杨梅等53种植物叶绿体基因组序列利用最大似然法构建系统发育树(图3),运行的最佳模型为GTR+F+I+G4。系统发育分析表明猴欢喜同日本杜英聚在一支,分支支持率达100%,属于杜英科。此外,猴欢喜与阳桃、红花酢浆草和Oxalis drummondii聚为一支,分支支持率为100%,说明猴欢喜同酢浆草科具有较近的亲缘关系。
3 讨论与结论
猴欢喜叶绿体基因组包含157 546 bp碱基,IR的长度为25 984 bp,GC含量为37.0%, 与被子植物叶绿体基因组已有数据(120 000 ~ 180 000 bp)相符(Wolf et al., 2010)。猴欢喜叶绿体基因组共注释了113个基因,与被子植物叶绿体基因组通常为120个左右的基因接近(杨芳,2019)。对比日本杜英的叶绿体基因结构,发现猴欢喜存在infA和ndhK两个基因。在被子植物中有部分物种的叶绿体基因组infA和ndh基因转移到核糖体,如朝鲜唐松草(Thalictrum ichangense)叶绿体基因组中infA基因转移至核糖体(Millen et al., 2001; Park & Jansen, 2015),蝴蝶兰(Phaiaenopsis aphrodite)中三个ndh基因(ndhA, ndhF和ndhH)转移到核糖体内(Chang et al., 2006)。此外,ndh基因在鞭寄生属(Hydnora)、菟丝子属(Cuscuta)等寄生植物和部分蕨类植物中缺失,可能是进化过程中适应水分过多环境的结果(DePamphilis & Palmer, 1990; McNeal et al., 2007; Wickett, 2008; Wicke et al., 2011; Kim, 2015; Naumann, 2016)。日本杜英在适应进化过程中叶绿体全基因组丢失了ndhK和infA两个基因,可能经历了不同的生境扩张。
SSR比较分析发现猴欢喜A/T碱基重复比G/C碱基重复多,SSR在非编码区比编码区更多,符合真核生物的SSR分布(Toth et al., 2000; Morgante et al., 2002; Zhao et al., 2014; Srivastava et al., 2019)。猴欢喜A/T碱基重复较多可能与减数分裂的重组热点有关(Heissl et al., 2018)。在基因组中,GC含量越高则DNA密度越大,基因序列越稳定。猴欢喜IR区的GC含量为42.9%,SSC区的GC含量较LSC区和IR区低,推测其SSC区有更多的SSR突变位点(Ohme-Takagi et al., 2000)。不同物种的叶绿体基因组ycf基因通常存在高频率的SSR,造成基因的高度变异,例如在爵床属(Justicia)中出现多态性位点次数最多的是ycf1基因(Kim & Lee, 2004;钮峥洋, 2020)。在猴欢喜叶绿体基因组的编码区中,ycf3基因的多态性位点最多,存在T和A两种重复类型,基因变异的几率大。总体来看,猴欢喜的叶绿体SSR呈现出分布不均匀、具有丰富变异性的特点,这些SSR可以作为猴欢喜属植物种内遗传变异和物种鉴定的微卫星分子标记。因此,猴欢喜叶绿体基因组的发表有助于杜英科植物的QTL分析、親缘关系鉴定和遗传多样性研究(刘列钊和林呐,2004)。
有研究發现叶绿体基因组的大小不同会导致IR/SC边界区出现扩张或者收缩现象,而IR/SC边界的扩张与收缩又与植物进化有密切的关系。IR边界区大的扩张或收缩可能与双链DNA的断裂修复有关,小的扩张或收缩与基因转化有关(Kim & Lee, 2004;Khakhlova & Bock, 2006;Hansen et al., 2007;Wang et al., 2008;Ma et al., 2013;梁凤萍等,2018;马丽,2020)。通过比较酢浆草目5种植物叶绿体全基因组的IR边界区,发现猴欢喜在IR/SC边界区存在较大的扩张,这可能与双链DNA的断裂修复有关;并且其边界区域与日本杜英相比,反而同酢浆草科的边界区域更加相似,可能反映猴欢喜属与酢浆草科的起源时间更接近,经历了相似的地质进化事件。对比IR边界区,猴欢喜的rps19基因横跨LSC/IRb区,而日本杜英则是rps3基因。研究结果表明rps19、 rps3基因在单、 双子叶植物叶绿体基因组中存在一定差异,在部分双子叶植物中rps19基因仅部分存在于IR区,而rps3基因横跨LSC/IRb区的现象仅在重楼属(Paris,藜芦科)和黑药花科(Melanthiaceae)植物中发现(Lin et al., 2012;Sarah et al., 2013;杨丽芳, 2019)。由此可见,杜英科的猴欢喜属与杜英属向东亚扩散的过程中经历了完全不同的复杂进化事件。
构建的系统发育树表明猴欢喜同日本杜英聚为一支,同属于杜英科酢浆草目,与之前Heibl & Renner (2012)和Magallon等(2015)构建的系统发育树相吻合。酢浆草目、卫矛目与金虎尾目聚为COM分支,与APG IV系统一致(Chase et al., 2016)。COM分支又与固氮分支(壳斗目、葫芦目、蔷薇目、豆目)同属于豆类分支(Fabids),这一结果进一步确定了COM分支的系统位置,也印证了叶绿体基因组系统发育分析方法在系统发育分析中的重要作用。
参考文献:
AMIRYOUSEFI A, HYVNEN J, POCZAI P, 2018. IRscope: An online program to visualize the junction sites of chloroplast genomes [J]. Bioinformatics, 34(17): 3030-3031.
BHATI R, KUMAR S, PAREKH MJ, 2018. Development of genomic simple sequence repeat (gSSR) markers in cumin and their application in diversity analyses and cross-transferability [J]. Ind Crop Product, 111(1): 158-164.
BRADFORD JC, BARNES RW, 2001. Phylogenetics and classification of Cunoniaceae (Oxalidales) using chloroplast DNA sequences and morphology [J]. Syst Bot, 26(2): 354-385.
CHANG CC, LIN HC, LIN L, et al.,2006. The chloroplast genome of Phalaenopsis aphrodite(Orchidaceae): comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications [J]. Mol Biol Evol, 23(2): 279-291.
CHANG HT(ZHANG HD), 1989. Flora Reipublicae Popularis Sinicae [M]. Beijing: Science Press, 49: 1-46. [张宏达, 1989. 中国植物志 [M]. 北京: 科学出版社, 49: 1-46.]
CHASE MW, CHRISTENHUSZ MJ, FAY MF, et al., 2016. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV [J]. Bot J Linn Soc,181(1): 1-20.
CHENG H, LI J, ZHANG H, et al.,2017. The complete chloroplast genome sequence of strawberry (Fragaria×ananassa Duch.) and comparison with related species of Rosaceae [J]. Peer J, 5(10): e3919-e3945.
COCK PJA, FIELDS CJ, GOTO N, et al.,2010. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants [J]. Nucl Acid Res, 38(6): 1767-1771.
CRAYN DM, DARREN M, ROSSETTO, et al.,2006. Molecular phylogeny and dating reveals an Oligo-Miocene radiation of dry-adapted shrubs (former Tremandraceae) from rainforest tree progenitors (Elaeocarpaceae) in Australia [J]. Amer J Bot, 93(9): 1328-1342.
DEPAMPHILIS CW, PALMER JD, 1990. Loss of photosynthetic and chlororespiratory genes from the plastid genome of a parasitic flowering plant [J]. Nature, 348(6299): 337-339.
HANSEN DR, DASTIDAR SG, CAI Z, et al., 2007. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae) [J]. Mol Phylogenet Evol, 45(2): 547-563.
HANSEN KD, BRENNER SE, DUDOIT S, 2010. Biases in Illumina transcriptome sequencing caused by random hexamer priming [J]. Nucl Acid Res, 38(12): e131-e137.
HEIBL C, RENNER SS, 2012. Distribution models and a date phylogeny for chilean Oxalis species reveal occupation of new habits by different lineages, not rapid adaptive radiation [J]. Syst Biol, 61(5): 823-834.
HEISSL A, BETANCOURT AJ, HERMANN P, et al.,2018. Length asymmetry and heterozygosity strongly influences the evolution of poly-A microsatellites at meiotic recombination hotspots [J]. BioRxiv, 18(1): 431841-431871.
JEON JH, KIM SC, 2019. Comparative analysis of the complete chloroplast genome sequences of three closely related east-Asian wild roses (Rosa sect. Synstylae; Rosaceae) [J]. Genes, 10(1): 23-36.
KEARSE M, MOIR R, WILSON A, et al.,2012. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data [J]. Bioinformatics, 28(12): 1647-1649.
KHAKHLOVA O, BOCK R, 2006. Elimination of deleterious mutations in plastid genomes by gene conversion [J]. Plant J, 46(1): 85-94.
KIM HT, 2015. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries [J]. PLoS ONE, 10(11): e0142215-e014232.
KIM KJ, LEE HL, 2004. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants [J]. DNA Res, 11(4): 247-261.
KUMAR S, STECHER G, TAMURA K, 2016. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets [J]. Mol Biol Evol, 33(7): 1870-1874.
LI D, ZHOU X, LI D, 2009. Extraction of hyperoside from Hypericum perforatum L. using CTAB reversed micelles [J]. Can J Chem Eng, 87(4): 584-590.
LI YT, ZHANG J, LI LF, GAO LJ, et al., 2018. Structural and comparative analysis of the complete chloroplast genome of Pyrus hopeiensis—“wild plants with a tiny population”—and three other Pyrus species [J]. Int J Mol Sci, 19(10): 3262-3280.
LIANG FP, WEN XN, GAO HY, et al., 2018. Analysis of chloroplast genomes features of Asteraceae species [J]. Genom Appl Biol, 37(12): 5437-5447. [梁凤萍, 文祥宁, 高赫一, 等, 2018. 菊科植物叶绿体基因组特征分析 [J]. 基因组学与应用生物学, 37(12): 5437-5447.]
LIN CP, WU CS, HUANG YY, et al., 2012. The complete chloroplast genome of Ginkgo biloba reveals the mechanism of inverted repeat contraction [J]. Genome Bio Evol, 4(3): 374-381.
LIU H, HE J, DING C, et al., 2018. Comparative analysis of complete chloroplast genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica revealing structural variations among genera in tribe Anemoneae (Ranunculaceae) [J]. Front Plant Sci, 9(1): 1097-1112.
LIU LZ, LIN N, 2004. Research advance of SSR(simple sequence repeat) in canola [J]. Chin Bull Life Sci, 3(1): 173-176. [劉列钊, 林呐, 2004. 油菜简单重复序列SSR(simple sequence repeat)研究进展 [J]. 生命科学, 3(1): 173-176.]
MA J, YANG B, ZHU W, et al., 2013. The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms [J]. Gene, 528(2): 120-131.
MA L, 2020. Comparative chloroplast genomics and DNA barcoding of Lagerstroemia species [D]. Hangzhou: Zhejiang A & F University: 26-27. [马丽, 2020. 紫薇属叶绿体比较基因组学及DNA条形码研究 [D]. 杭州: 浙江农林大学: 26-27.]
MAGALLON S,GMEZ-ACEVEDO S, REYES LLS, et al., 2015. A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity [J]. New Phytol, 207(2): 437-453.
MARECHAL A, BRISSON N, 2010. Recombination and the maintenance of plant organelle genome stability [J].New Phytol, 186(2): 299-317.
MCNEAL JR, KUEHL JV, BOORE JL, et al., 2007. Complete plastid genome sequences suggest strong selection for retention of photosynthetic genes in the parasitic plant genus Cuscuta [J]. BMC Plant Biol, 7(1): 57-78.
MILLEN RS, OLMSTEAD RG, ADAMS KL, et al., 2001. Many parallel losses of infA from chloroplast angiosperm evolution with multiple independent transfers to the nucleus [J]. Plant Cell, 13(3): 645-658.
MORGANTE M, HANAFEY M, POWELL W, 2002. Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes [J]. Nat Genet, 30(1): 194-200.
NAUMANN J, 2016. Detecting and characterizing the highly divergent plastid genome of the nonphotosynthetic parasitic plant Hydnora visseri (Hydnoraceae) [J]. Genome Biol Evol, 8(2): 345-363.
NIU ZY, 2020. Phylogenomics of Asian Justicia L. based on complete chloroplast genomes [D]. Nanjing: Nanjing Forestry University: 61-65. [钮峥洋, 2020. 亚洲广义爵床属叶绿体系统发育基因组学研究 [D]. 南京: 南京林业大学: 61-65.]
OHME-TAKAGI M, SUZUKI K, SHINSHI H, 2000. Regulation of ethylene-induced transcription of defense genes [J]. Plant Cell Physiol, 41(11): 1187-1192.
PALMER JD, 1985. Comparative organization of chloroplast genomes [J]. Ann Rev Genet, 19(1): 325-354.
PARK S, JANSEN RK, 2015. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae [J]. BMC Plant Biol, 15(1): 40-52.
PHOON SN, 2015. Systematics and biogeography of Elaeocarpus (Elaeocarpaceae) [D]. Townsville: James Cook University: 82-85.
POGSON BJ, GANGULY D, ALBRECHT-BORTH V, 1847. Insights into chloroplast biogenesis and development [J]. BBA-Bioenergetics, 1847(9): 1017-1024.
PRICE MN, DEHAL PS, ARKIN AP, 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix [J]. Mol Biol Evol, 26(7): 1641-1650.
PRJIBELSKI A, ANTIPOV D, MELESHKO D, et al., 2020. Using SPAdes de novo assembler [J]. Curr Protoc Bioinform, 70(1): e102-e130.
RAMAN G, PARK S, 2015. Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. longicalyncinus, from a comparative genomics perspective [J]. PLoS ONE, 10(10): e0141329-e0141349.
SAHU J, SARMAH R, DEHURY B, et al., 2012. Mining for SSRs and FDMs from expressed sequence tags of Camellia sinensis [J]. Bioinformation, 8(6): 260-266.
SARAH S, KIM JH, JUNG SK, et al., 2013. Complete chloroplast genome of Chionographis japonica (Willd.) Maxim. (Melanthiaceae): comparative genomics and evaluation of universal primers for Liliales [J]. Plant Mol Biol Rep, 31(6): 1407-1421.
SHINOZAKI K, OHME M, TANAKA M, et al., 1986. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression [J]. EMBO J, 5(9): 2029-2043.
SRIVASTAVA S, AVVARU AK, SOWPATI DT, et al., 2019. Patterns of microsatellite distribution across eukaryotic genomes [J]. BMC Genomics, 20(1): 153-166.
SU N, HE ZF, OU PH, et al., 2020. Comparative analysis of the chloroplast genome in Triticum species [J]. J Tritic Crops, 40(1): 55-64. [蘇宁, 何兆峰, 欧平和, 等, 2020.小麦属植物叶绿体基因组结构的比较分析 [J].麦类作物学报, 40(1):55-64.]
TAKHTAJAN AL, 1980. Outline of the classification of flowering plants (Magnoliophyta) [J]. Bot Rev, 46(3): 225-359.
TAO XL, MA LC, NIE B, et al., 2017. The draft and characterization of the complete chloroplast genome of Vicia sativa cv. Lanjian No.3 [J]. Prat Sci, 34(2): 321-330. [陶晓丽, 马利超, 聂斌, 等, 2017. ‘兰箭3号’春箭筈豌豆叶绿体全基因组草图及特征分析 [J]. 草业科学, 34(2): 321-330.]
THORNE RF, 2000. The classification and geography of the flowering plants: Dicotyledons of the class Angiospermae [J]. Bot Rev, 66(4): 441-647.
TOTH G, GASPARI Z, JURKA J, 2000. Microsatellites in different eukaryotic genomes survey and analysis [J]. Genome Res, 10(7): 967-981.
WANG RJ, CHENG CL, CHANG CC, et al., 2008. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots [J]. BMC Evol Biol, 8(1): 36-49.
WICKE S, SCHNEEWEISS GM, DEPAMPHILIS CW, et al., 2011. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function [J]. Plant Mol Biol, 76(3): 273-297.
WICKETT NJ, 2008. Functional gene losses occur with minimal size reduction in the plastid genome of the parasitic liverwort Aneura mirabilis [J]. Mol Biol Evol, 25(2): 393-401.
WOLF PG, DER J, DUFFY A, et al., 2010. The evolution of chloroplast genes and genomes in ferns [J]. Plant Mol Biol, 76(3): 251-261.
XIE YF, 2018. Ataxonomic study of Elaeocarpaceae in East Asia and the Pan-Himalayas [D]. Beijing: Beijing Forestry University: 14-15. [謝宜飞, 2018.东亚及泛喜马拉雅地区杜英科的分类研究 [D]. 北京: 北京林业大学: 14-15.]
YANG F, 2019. Sequencing andstuctural analysis of chloroplast genome in Rosa banksias [J]. Genom Appl Biol, 38(8): 3586-3594. [杨芳, 2019. 七里香蔷薇叶绿体基因组测序及结构分析 [J]. 基因组学与应用生物学, 38(8): 3586-3594.]
YANG LF, 2019. Plastid phyogenomics and biogeography of Paris [D]. Kunming: Yunnan University: 44-45. [杨丽芳, 2019. 重楼属叶绿体系统发育基因组学与生物地理学研究 [D]. 昆明: 云南大学: 44-45.]
YANG YM, JIAO J, FAN XC, et al., 2019. Complete chloroplast genome sequence and characteristics analysis of Vitis ficifolia [J]. Acta Hort Sin, 46(4): 635-648. [杨亚蒙, 焦健, 樊秀彩, 等, 2019. 桑叶葡萄叶绿体基因组及其特征分析 [J]. 园艺学报, 46(4): 635-648.]
ZHANG D, GAO F, JAKOVLI I, et al., 2020. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies [J]. Mol Ecol Resour, 20(1): 348-355.
ZHANG X, RONG C, QIN L, et al., 2018. Complete chloroplast genome sequence of Malus hupehensis: genome structure, comparative analysis, and phylogenetic relationships [J]. Molecules, 23(11): 2917-2929.
ZHANG X, ZHOU T, KANWAL N, et al., 2017. Completion of eight gynostemma bl. (cucurbitaceae) chloroplast genomes: characterization, comparative analysis, and phylogenetic relationships [J]. Front Plant Sci, 8(1): 1583-1595.
ZHAO ZX, GUO C, SUTHARZAN S, et al., 2014. Genome-wide analysis of tandem repeats in plants and green algae [J]. G3 Genes, 4(1): 67-78.
ZHENG SY, POCZAI P, HYVNENJ, et al., 2020. Chloroplot: an online program for the versatile plotting of organelle genomes [J]. Front Genet, 11(1): 1-8.
(责任编辑 何永艳)
1943501186371
