
响应面法优化胡黄连苷Ⅱ硅胶柱色谱纯化工艺△
2022-03-16 06:07:26毕艳艳郝大伟a代立平唐伟铭龙瑞沈庆国
中国现代中药 2022年2期
毕艳艳,郝大伟a,代立平,唐伟铭,龙瑞,沈庆国*
1.鲁南厚普制药有限公司,山东 临沂 276006;
2.中药制药共性技术国家重点实验室,山东 临沂 276006
胡黄连是玄参科植物胡黄连Picrorhiza scrophulariifloraPennell 的干燥根茎,主要分布于我国西藏、云南和四川的高原地区。近年来,国内外的研究发现,胡黄连中有效成分主要为环烯醚萜苷(胡黄连苷)类、葫芦素类及酚苷类,其具有保肝利胆、抗肿瘤、抗糖尿病等多种功效[1]。胡黄连苷以胡黄连苷Ⅰ、胡黄连苷Ⅱ为主要有效成分,《中华人民共和国药典》2020 年版相关内容规定,西藏胡黄连中胡黄连苷Ⅰ和胡黄连苷Ⅱ的总量不得少于9.0%[2];其中,胡黄连苷Ⅱ含量最高,为最主要成分,且具有广泛药理活性,对肝脏、大脑、心脏、肾脏有显著的保护作用,具有抗氧化、抗炎、平喘等功效[3-4]。
硅胶柱色谱是天然产物分离最常用的方法之一,但常规装柱采用常压敞口色谱柱,具有柱床松散、样品载样量小、重现性差、分离速度慢、生产周期长、使用溶剂多、有机溶剂挥发污染环境等缺点,而高压色谱柱能避免常压色谱柱的不足,已被广泛用于食品、医药、农业等领域[5-7]。本实验引入高压硅胶色谱柱的中低压色谱技术,运用Box-Behnken响应面法设计原理,采用薄层色谱和高效液相色谱法(HPLC)进行定性定量分析,以胡黄连苷Ⅱ质量分数、回收率为评价指标,探讨硅胶柱色谱纯化胡黄连苷Ⅱ最佳工艺条件,为胡黄连苷Ⅱ开发利用提供参考。
1 材料
1.1 仪器
DF-60-A 型水冷双级高速齿轮传动粉碎机(温岭市林大机械有限公司);1260 型高效液相色谱仪(Agilent公司);R-300 型旋转蒸发器(瑞士Buchi公司);DLSK-10/30 型低温冷却液循环泵(郑州科泰试验设备有限公司);XS204 型电子分析天平(Mettler-Toledo公司);Prep200型高压制备液相系统(上海赛梵科分离技术有限公司);DAC-50 型上样柱(上海赛梵科分离技术有限公司,650 mm);DAC-50 型色谱柱(上海赛梵科分离技术有限公司,1300 mm)。
1.2 试药
对照品胡黄连苷Ⅰ(批号:111727-201702,纯度:95.6%);胡黄连苷Ⅱ(批号:111596-201805,纯度:93.4%)均购自中国食品药品检定研究院;三氯甲烷(金岭化工股份有限公司,批号:20190219,工业级);硅胶300~400 目(青岛海洋化工有限公司,批号:19021030333);甲醇(兖矿国宏化工有限公司,批号:20191218,工业级);高效液相用试剂均为色谱纯;分析用试剂均为分析纯。
胡黄连(产地为西藏自治区,批号:190401)经中药制药共性技术国家重点实验室范建伟高级工程师鉴定为玄参科植物胡黄连Picrorhiza scrophulariifloraPennell的干燥根茎。
2 方法
2.1 胡黄连苷Ⅱ质量分数测定
2.1.1 HPLC 色谱条件 色谱柱Agilent ZORBAX SB C18(250 mm×4.6 mm,5 μm);流动相:甲醇-水-磷酸(35.0∶65.0∶0.1);流速:1.0 mL·min-1;柱温:30 ℃;检测波长:275 nm;进样量:10 μL。
2.1.2 对照品溶液的制备 精密称取胡黄连苷Ⅱ对照品适量,加甲醇制成含胡黄连苷Ⅱ0.268 mg·mL-1的溶液,即得对照品储备液。精密吸取对照品储备液5 mL 置于20 mL 量瓶中,加甲醇稀释定容至至刻度,摇匀,即得。
2.1.3 供试品溶液的制备 精密称取各供试样品30 mg置于50 mL量瓶中,加甲醇超声溶解,冷却并定容至刻度,摇匀,得供试品储备液。精密吸取供试品储备液5 mL 置于25 mL 量瓶中,加甲醇稀释定容至刻度,摇匀,即得。
2.1.4 绘制标准曲线 分别精密移取2.1.2 项下对照品溶液1、2、4、5、8、10 mL置于20 mL量瓶中,加甲醇溶解定容至刻度,按2.1.1项下条件进样测定,记录峰面积值。以峰面积为纵坐标(Y),对照品质量浓度为横坐标(X),绘制标准曲线,并计算线性回归方程,胡黄连苷Ⅱ:Y=14 281 948.778X-13 408.559(r=0.999),线性范围为0.013 4~0.134 0 mg·mL-1。
2.2 拌样硅胶制备
称取定量已知质量分数的胡黄连粗粉,按照料液比为1∶20加入95%乙醇,提取3次,提取总时间为4.5 h,合并提取液,滤过,浓缩至适量,得胡黄连浸膏[8-10]。按药材-硅胶质量比为3∶1称取300~400目硅胶,与胡黄连浸膏混匀拌样,干燥粉碎,得拌样硅胶,采用HPLC测定胡黄连苷Ⅱ质量分数。
2.3 硅胶柱色谱
称取定量300~400 目硅胶,加入流动相三氯甲烷-甲醇,超声30 min,将硅胶匀浆液缓慢加入色谱柱中,开启气压,缓慢压实,启动流动相泵平衡色谱柱。
称取2.2项下制备的拌样硅胶,加入流动相三氯甲烷-甲醇,超声30 min,将拌样硅胶匀浆液缓慢加入上样柱中,将上样柱与色谱柱串联,开启气压,缓慢压实。启动流动相泵进行洗脱,同时采集色谱图,待目标峰出现后,收集洗脱液,每30 min 收集一段,蒸干各段洗脱液,采用HPLC 测定胡黄连苷Ⅱ质量分数,去除峰前峰后质量分数较低的各段,合并质量分数较高的各段,得目标产品(质量分数>40%)。通过公式(1)计算胡黄连苷Ⅱ的回收率。

式中,R为胡黄连苷Ⅱ的回收率;m1为产品的质量;w1为产品中胡黄连苷Ⅱ的质量分数;m2为拌样硅胶的质量;w2为拌样硅胶中胡黄连苷Ⅱ的质量分数。
2.4 单因素试验
2.4.1 洗脱剂三氯甲烷-甲醇比例的确定 根据相关文献报道及前期预试验,兼顾溶剂之间的极性、后续溶剂回收利用,本实验选取三氯甲烷-甲醇的溶剂体系作为洗脱剂[11-12]。
洗脱剂对目标产物的洗脱分离效果决定了目标产物的质量分数和回收率。本实验采用薄层色谱法对胡黄连苷Ⅱ的洗脱剂三氯甲烷-甲醇比例进行考察。吸取同体积样品和对照品点样在同一硅胶板上(5份),待溶剂挥干后,分别放置到已配置饱和的不同比例洗脱剂的展缸中,洗脱剂三氯甲烷-甲醇比例分别为4∶1、5∶1、6∶1、7∶1、8∶1,待洗脱剂展开至10 cm 左右取出,挥干洗脱剂,显色,观察。计算目标产物在不同洗脱剂比例中比移值(Rf)。
2.4.2 色谱硅胶-上样样品(原药材)质量比的确定 按照色谱硅胶-上样样品(原药材)质量比为1∶1、1.4∶1.0、1.7∶1.0、2∶1、2.4∶1.0 分别称取按照2.2 项下方法制备的拌样硅胶、色谱硅胶装柱,经三氯甲烷-甲醇比例为6∶1进行洗脱,洗脱流速为6 mL·min-1,分段收集,蒸干各段洗脱液,采用HPLC 测定胡黄连苷Ⅱ质量分数,计算最终产品胡黄连苷Ⅱ质量分数、回收率。
2.4.3 洗脱流速的确定 按照色谱硅胶-上样样品(原药材)质量比为1.7∶1.0分别称取按照2.2项下制备拌样硅胶、色谱硅胶装柱,经三氯甲烷-甲醇(6∶1)进行洗脱,洗脱流速分别为4、6、7、8、10 mL·min-1,分段收集,蒸干各段洗脱液,采用HPLC 测定胡黄连苷Ⅱ质量分数,计算最终产品胡黄连苷Ⅱ质量分数及回收率。
2.5 响应面试验
在单因素试验的基础上,根据响应面法设计原理[7,13-14],以洗脱剂三氯甲烷-甲醇比例(A)、色谱硅胶-上样样品(原药材)质量比(B)、洗脱流速(C)为响应因素,以胡黄连苷Ⅱ质量分数(R1)、回收率(R2)、综合评分(R综)为响应值,进行三因素三水平的响应面分析试验,设计了17 个试验点,筛选各因素的最优条件。
因胡黄连苷Ⅱ的质量分数和回收率具有相关性,质量分数增大会导致其回收率的降低,反之亦然。在保证胡黄连苷Ⅱ质量分数>40%的前提下,尽可能提高其回收率,因此,胡黄连苷Ⅱ质量分数和回收率2个指标所占权重分别为60%、40%,其加权后的综合评分为质量分数和回收率2 个指标得分之和。胡黄连苷Ⅱ柱色谱纯化工艺综合评价指标见表1。
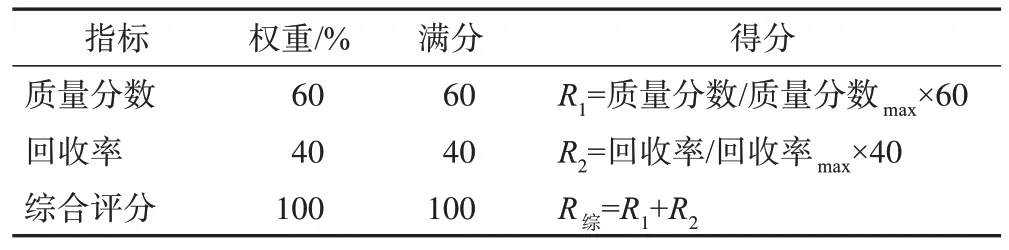
表1 胡黄连苷Ⅱ柱色谱纯化工艺综合评价指标
3 结果与讨论
3.1 单因素试验结果
3.1.1 洗脱剂三氯甲烷-甲醇比例的确定 洗脱剂三氯甲烷-甲醇比例分别为4∶1、5∶1、6∶1、7∶1、8∶1,胡黄连苷Ⅱ展开的Rf见表2,洗脱剂比例小,洗脱剂极性较强,化合物移动的速度越快,洗脱分离效果较差;洗脱剂比例大,洗脱剂极性低,洗脱所需的时间长,消耗洗脱剂增多,且三氯甲烷毒性较大,生产成本提高。因此,选择洗脱剂三氯甲烷-甲醇5∶1、6∶1、7∶1为响应因素考察范围。

表2 不同洗脱剂比例胡黄连苷Ⅱ展开的Rf
3.1.2 色谱硅胶-上样样品(原药材)质量比的确定 按照色谱硅胶-上样样品(原药材)质量比为1∶1、1.4∶1.0、1.7∶1.0、2∶1、2.4∶1.0 称取硅胶装柱,胡黄连苷Ⅱ质量分数和回收率结果见表3。

表3 不同色谱硅胶-上样样品(原药材)质量比胡黄连苷Ⅱ质量分数和回收率
结合生产需要,为得到质量分数较高目标产品,当上样样品质量已定,色谱硅胶-上样样品(原药材)质量比小时,色谱硅胶用量少,会出现超载,造成分离纯化效果差,使胡黄连苷Ⅱ回收率较低;随着色谱硅胶-上样样品(原药材)质量比增大,色谱硅胶用量增多,柱径高比增加,胡黄连苷Ⅱ质量分数不断增大,回收率也增大;但色谱硅胶用量太多,会引起有效成分残存在硅胶上,使实际回收率下降,且生产成本会提高。综合考虑,选择色谱硅胶-上样样品(原药材)质量比1.4∶1.0、1.7∶1.0、2∶1 为响应因素考察范围。
3.1.3 洗脱流速的确定 经三氯甲烷-甲醇洗脱,洗脱流速分别为4、6、7、8、10 mL·min-1,胡黄连苷Ⅱ质量分数和回收率结果见表4。
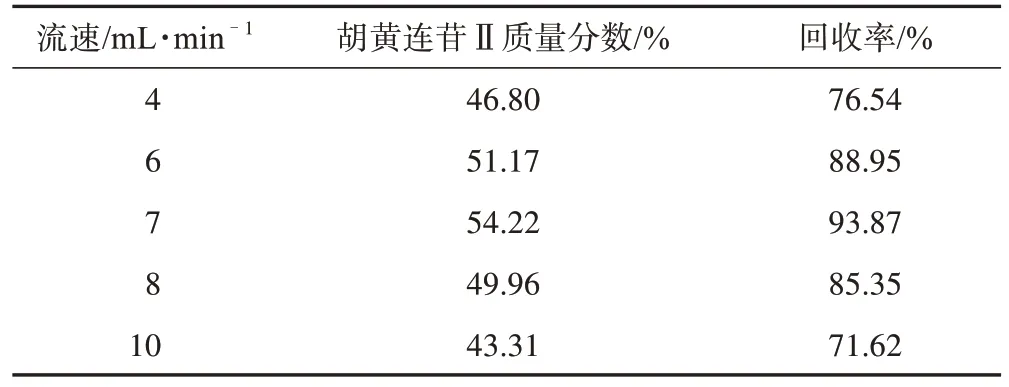
表4 不同洗脱流速胡黄连苷Ⅱ质量分数和回收率
当洗脱流速太低时,导致轴向扩散,使柱色谱分离纯化效果变差,胡黄连苷Ⅱ质量分数、回收率较低,且所需要的时间长;当洗脱速度太大时,两相间的浓度难以分配平衡,洗脱剂未能对被洗脱物进行充分洗脱,造成胡黄连苷Ⅱ质量分数、回收率也较低,且消耗洗脱剂会增多。结合生产需要,为得到质量分数较高目标产品,综合考虑选择洗脱流速为6、7、8 mL·min-1为响应因素考察范围。
3.2 响应面试验结果
在单因素试验的基础上,响应面优化胡黄连苷Ⅱ柱色谱纯化工艺的因素水平设计见表5,Box-Behnken响应面优化胡黄连苷Ⅱ柱色谱纯化工艺方案设计及结果见表6,Box-Behnken响应面优化胡黄连苷Ⅱ柱色谱纯化工艺二次回归方程方差分析结果见表7。

表5 响应面优化胡黄连苷Ⅱ柱色谱纯化工艺的因素水平设计
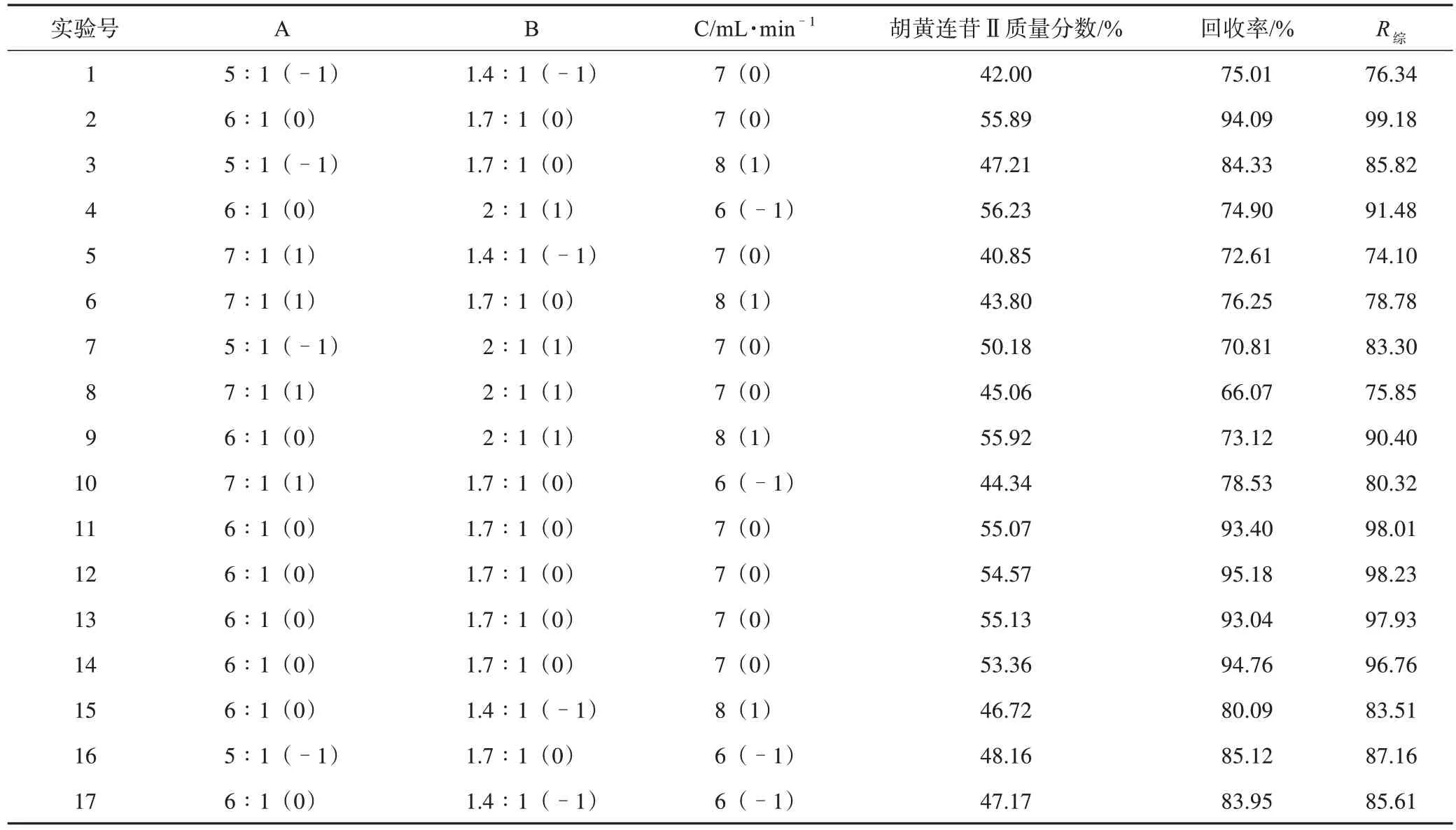
表6 Box-Behnken响应面优化胡黄连苷Ⅱ柱色谱纯化工艺设计方案及结果
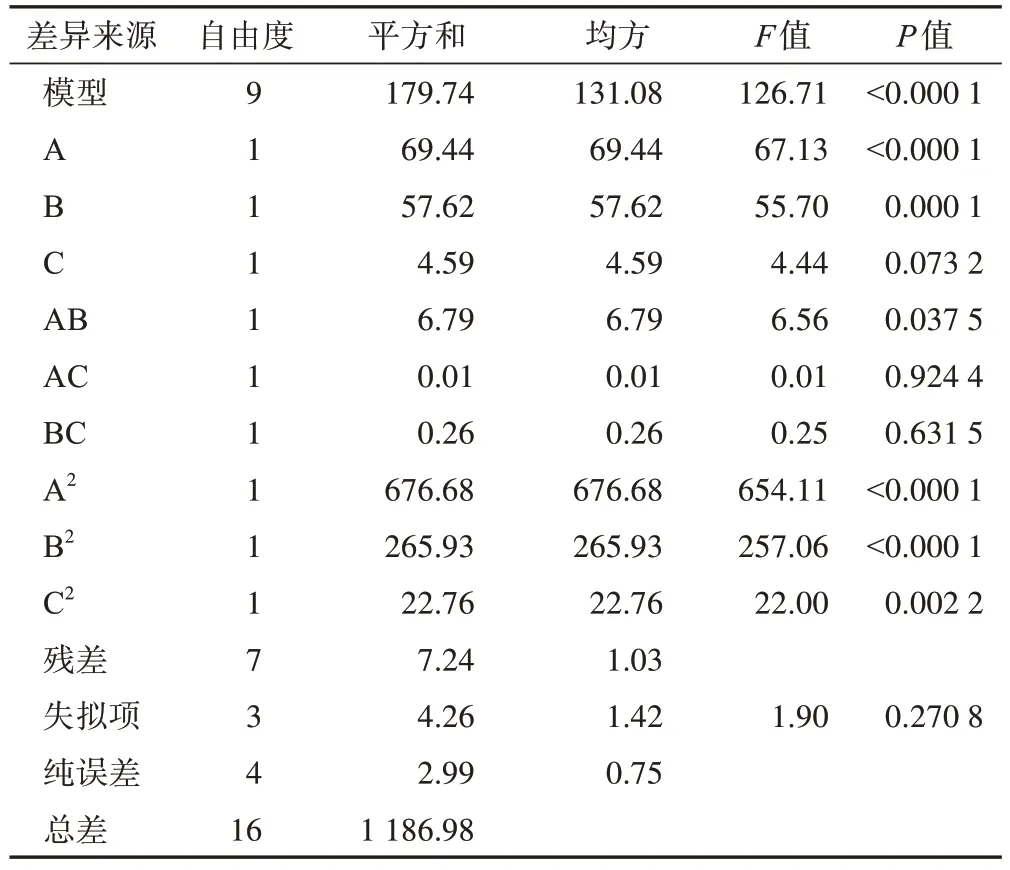
表7 Box-Behnken响应面优化胡黄连苷Ⅱ柱色谱纯化工艺二次回归方程方差分析结果
利用Design-Expert v 8.0.6 软件对表6 中的数据进行二次多元回归拟合,R综对各因素的二次多项回归模型方程为R综=-755.96+156.91A+329.27B+30.64C-4.34AB-0.50AC+0.85BC-12.68A2-88.30B2-2.32C2,决定系数R2=0.993 9。
由表7 结果,对R综,回归模型P<0.01,说明回归模型极显著,模型成立;A、B、A2、B2、C2因素P<0.01,交互项AB的P<0.05,差异有统计学意义;失拟项P>0.05,说明失拟项差异无统计学意义,提示回归方程拟合度良好,可用于分析与预测。各因素对R综值的影响大小顺序为A>B>C。
3.3 响应面模型分析
利用Design-Expert v 8.0.6软件得到二次回归方程的响应面图,以此评价实验各因素之间的交互强度,以确定最佳纯化工艺参数。结果见图1~3。
等高线越接近椭圆形或响应面线越陡峭说明这2 种交互因素对响应值影响越显著,反之,不显著。由图1~3 可知,色谱硅胶-上样样品质量比和洗脱剂比例对R综的影响差异有统计学意义,洗脱剂比例和洗脱流速及色谱硅胶-上样样品质量比和洗脱流速对R综的影响差异无统计学意义。
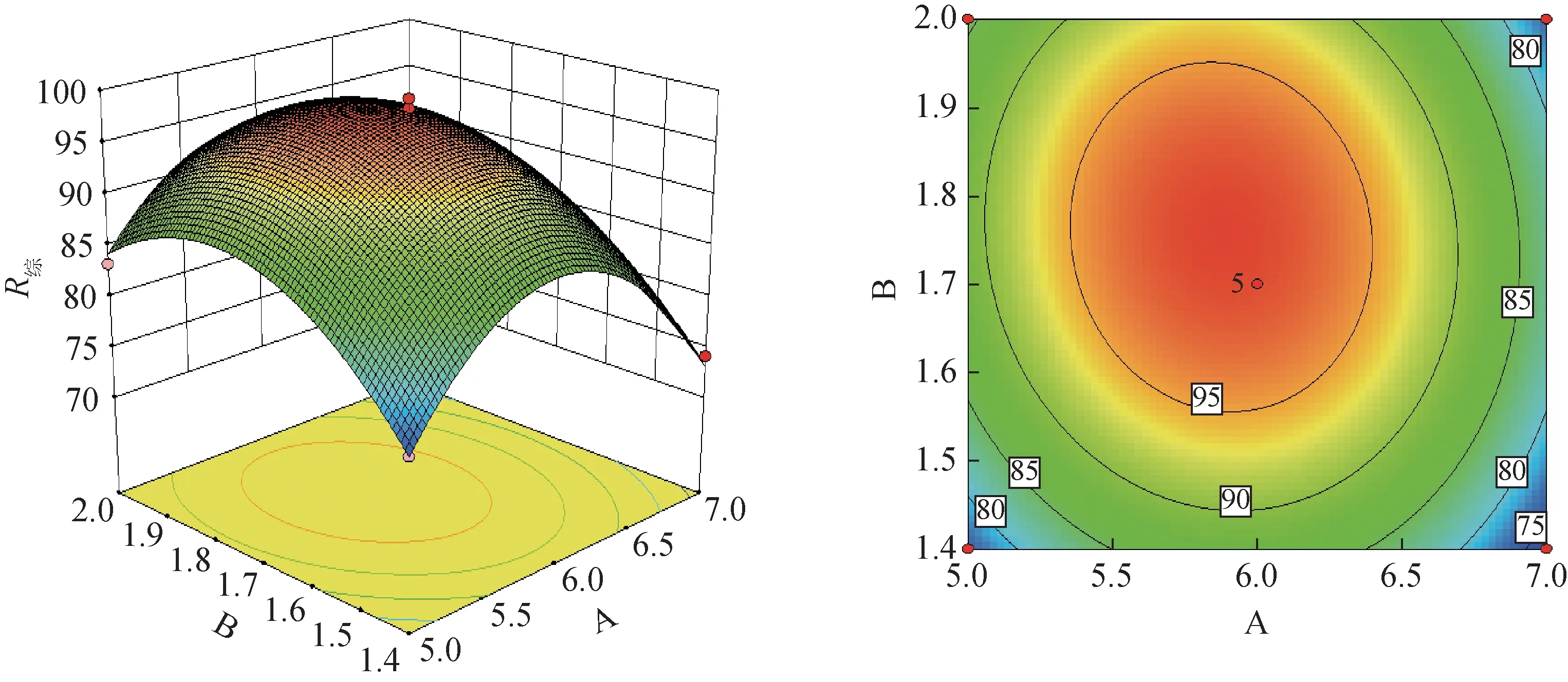
图1 色谱硅胶-上样样品质量比和洗脱剂比例对R综的响应面和等高线图

图2 洗脱剂比例和洗脱流速对R综的响应面和等高线图
利用Design-Expert v 8.0.6软件优化得到最佳柱色谱纯化工艺∶洗脱剂三氯甲烷-甲醇为5.88∶1.00,色谱硅胶-上样样品(原药材)质量比为1.74∶1.00,洗脱流速为6.85 mL·min-1,胡黄连苷Ⅱ质量分数为55.41%,回收率为93.65%,R综为98.48。结合实际所需,最终确定最佳工艺:洗脱剂三氯甲烷-甲醇为6∶1,色谱硅胶-上样样品(原药材)质量比为1.7∶1.0,洗脱流速为7 mL·min-1。
3.4 验证实验
按照上述最佳工艺条件:洗脱剂三氯甲烷-甲醇比例为6∶1,色谱硅胶-上样样品(原药材)质量比为1.7∶1.0,洗脱流速为7 mL·min-1,完成3 批验证实验,得到胡黄连苷Ⅱ质量分数、回收率平均值分别为54.99%、93.59%,RSD 分别为1.33%、1.17%;R综平均值为98.62。结果提示,优化得到硅胶柱色谱纯化胡黄连苷Ⅱ工艺稳定可靠。
4 结论
本实验通过Box-Behnken 响应面法优化得到胡黄连苷Ⅱ硅胶柱色谱纯化最佳工艺:洗脱剂三氯甲烷-甲醇比例为6∶1,色谱硅胶-上样样品(原药材)质量比为1.7∶1.0,洗脱流速为7 mL·min-1。该方法操作简单、稳定性好、节省成本,胡黄连苷Ⅱ质量分数、回收率较高,为将来胡黄连苷Ⅱ作为原料药投入大生产提供一定参考价值。
猜你喜欢
净水技术(2023年11期)2023-11-20 09:43:44
中学生数理化·八年级物理人教版(2023年4期)2023-05-05 07:29:42
中学生数理化·八年级物理人教版(2022年4期)2022-04-26 14:11:16
食品安全导刊(2021年36期)2021-03-14 04:53:24
科学导报(2020年75期)2020-12-21 03:53:32
大众科学(2020年7期)2020-10-26 09:24:30
氯碱工业(2020年6期)2020-03-01 07:30:33
小天使·六年级语数英综合(2018年1期)2018-10-08 09:32:50
中成药(2017年4期)2017-05-17 06:09:46
核科学与工程(2015年3期)2015-09-26 11:58:24
