
多孔SiC纳米片的原位合成及光催化性能
2022-03-16 13:26朱子轩郑雨佳
无机化学学报 2022年3期
朱子轩 郑雨佳 王 丹 曹 宏,2 薛 俊*,,2
(1武汉工程大学材料科学与工程学院, 武汉 430205)
(2湖北省环境材料与膜技术工程技术研究中心,武汉 430205)
0 引 言
碳化硅(SiC)是最重要的半导体材料之一,可以表现出优异的性能,如可调宽带隙、高强度、高热导率、优异的抗热冲击性、低热膨胀系数和优异的化学惰性。因此,这种材料可以在高温和各种酸碱条件下保持稳定。这些独特的性能使这种材料成为大功率电子产品、恶劣环境电子产品、蓝光二极管、传感器、光催化剂和异质催化剂载体的理想候选材料。众所周知,SiC的微观结构可以显著影响其特性。SiC纳米材料具有尺寸小、比表面积大、表面原子比例高、表面活性高、量子效应和纳米效应等特点,有望在光催化制氢和超级电容器方面取得突破[1-2]。目前对SiC纳米材料的研究大多集中在纳米颗粒、纳米线和纳米纤维上。研究者已经开发了许多方法来制备这些SiC纳米材料,如溶胶-凝胶法、碳热还原法[3-5]、碳纳米管模板生长法[6]、化学气相沉积法[7]和水热及溶剂热法[8-9]。然而,关于多孔SiC纳米片(SiCNSs)的简单制备方法的报道很少[10]。
Houmad等通过使用全电位线性粘贴平面波法(FP-LAPW)计算了SiC纳米晶体的电子和光学特性,并得到了约2.2 eV的理论带隙宽度[11]。我们以可膨胀石墨为原料,通过高温膨胀和卧式砂磨机的砂磨,得到了石墨片。然后将SiO和Si粉与石墨片混合作为前驱体,通过碳热还原法合成了多孔的SiCNSs,其禁带宽度与Houmad等所制备的SiC相似,而制备方式更为简便且原料廉价、简单易得。
1 实验部分
1.1 试剂与仪器
主要试剂包括可膨胀石墨、SiO、Si粉、氢氟酸、硝酸、去离子水。主要仪器:砂磨机、管式气氛炉、德国布鲁克D8 ADVANCE BRUKER型X射线衍射仪(XRD,Cu Kα射线,λ=0.154 06 nm,工作电压40 kV,工作电流40 mA,扫描范围10°~80°,扫描速度8(°)·min-1)、日本电子株式会社JSM-6700场发射扫描电子显微镜(FESEM,工作电压5.0 kV)和JEM-2100F型透射电子显微镜(TEM,200 kV)、美国康塔克默NOVA2000型全自动比表面积及孔隙分析仪、日本岛津UV2600紫外可见分光光度计、中教金源-CELLB70光催化装置(500 W氙灯)。
1.2 碳化硅的制备
将氧化铝坩埚放入到高频感应加热机的加热源中,设置温度为1 000℃。将可膨胀石墨缓慢加入到氧化铝坩埚中进行高温膨化,待石墨剥离完毕后(5~10 min),关闭加热设备,冷却至室温后取出膨胀石墨。将制备的膨胀石墨(10 g)、聚氧乙烯醚(0.5 g)缓慢加入到去离子水(100 mL)中,混合均匀。将混合后的膨胀石墨浆料倒入砂磨机漏斗中,收集物料并记录砂磨循环1次所用时间。砂磨每循环1次,取样进行粒度测试,如果粒度变小,继续砂磨,直到粒度不变即得到石墨薄片,所需时间即为最大砂磨时间。石墨薄片粒度大小以D50表示,即一个样品的累计粒度分布百分数达到50%时所对应的粒径(中位径或中值粒径)。D50与砂磨时间关系如图1a所示,400 min为其最大砂磨时间,此后粒度无明显变化。如图1b所示,石墨薄片的D50的对数与砂磨时间呈线性关系。为研究不同粒径的石墨薄片对生成SiCNSs的性质及产氢性能的影响,通过中位数原理选取砂磨时间0、5、30、125和400 min的石墨薄片作为碳源,其粒径分别为72.26、42.21、18.75、11.43、2.34 µm(图2)。

图1 石墨薄片的D50与砂磨时间关系:(a)测试曲线;(b)拟合曲线Fig.1 Relationship between D50of graphite flake and sanding time:(a)observed curve;(b)fitted curve
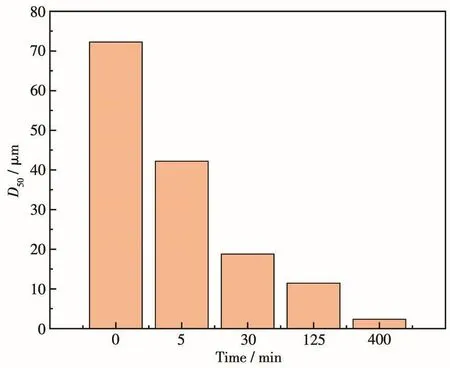
图2 不同时间砂磨石墨薄片的D50Fig.2 D50of graphite flakes sanded for different times
按照物质的量之比 nC∶nSiO∶nSi=2.5∶1∶1 分别称取膨胀石墨、SiO和Si硅粉置于烧杯中,加入去离子水制成固体含量为10%的浆料。随后倒入砂磨机漏斗中砂磨(0、5、30、125、400 min)混合均匀,砂磨后的浆料在磁力搅拌器中加热搅拌至糊状,放入100℃干燥箱干燥24 h得到前驱体。然后将前驱体放入氧化铝管式炉中焙烧,焙烧温度为1 350℃,保温时间2 h,冷却至室温后得到初始产物。将所得产物浸泡在10%的混合酸(VHF∶VHNO3=3∶1)溶液中,搅拌、浸泡12 h以除去硅的氧化物。其后的样品经水洗干燥后在650℃、空气气氛下焙烧4 h,除去初始产物中残余的石墨,最后得到纯的SiCNSs。流程图如图3所示。

图3 SiCNSs制备流程图Fig.3 Flow chart of preparation of SiCNSs
1.3 SiCNSs光解水产氢测试
在光催化产氢系统中,称取300 mg SiCNSs分散于200 mL去离子水中,光照之前,对反应器进行超声分散和抽真空处理(约30 min)以排除杂质气体。光源为300 W的氙灯,在氙灯上安装截止滤光片(λ≥420 nm)以得到波长420 nm以上的可见光。整个反应过程需要不断搅拌,并通入冷却循环水使反应温度保持在25℃。产生的气体用气相色谱仪(高纯N2作为载气)进行检测。
2 结果与讨论
2.1 XRD分析
图4为不同砂磨时间前驱体制备SiCNSs的XRD图。从图4中可以看到所有样品的XRD图基本一致,与PDF卡片(PDF No.29-1129)对照,位于35.6°、41.4°、60.0°和71.8°处的峰分别对应于立方相3C-SiC的(111)、(200)、(220)和(311)晶面。对XRD图进行精修后得出其晶格常数a=0.435 89 nm,这与标准图的晶格常数a=0.435 9 nm几乎一致。以上均表明合成的样品为3C-SiC,适合作为半导体光催化剂。此外,还可以看到随着砂磨时间的增加,样品衍射峰变尖锐且峰强度明显变大。这是因为随着砂磨时间的增加,反应物尺寸减小,高温下反应更完全。图中除了SiC的衍射峰外,并未出现其他物相的衍射峰,如SiO2、碳和其它不纯物,说明制得的是纯的3C-SiC。

图4 不同砂磨时间的前驱体制备的SiCNSs的XRD图Fig.4 XRD patterns of SiCNSs prepared with the precursors sanded for different times
2.2 形貌分析
产物的形貌通过FESEM作进一步分析。图5a~5e为不同砂磨时间前驱体制备的SiCNSs的FESEM图。从图中可以清楚地看到,在1 350℃下保温2 h得到的SiCNSs由多个纳米片堆积而成,其一次片层结构的平均粒径介于几十到几百纳米之间。反应前驱体砂磨0 min时粒径最大,通过测量可知其粒径为400~500 nm。而反应前驱体砂磨400 min时粒径最小,产物由许多细小、均匀的颗粒以及少量的纳米线通过相互黏结、交叉,形成了絮状结构。从前文可知,采用机械砂磨的方式破坏了石墨的层状结构,得到了石墨薄片,而400 min为其砂磨极限,此后粒径变化不大,此时的石墨薄片几乎可以近似看作石墨颗粒。由此,我们推测产物SiCNSs在很大程度上继承了石墨薄片的结构,石墨薄片不仅作为碳源参与反应,而且还为SiCNSs的生长提供了结构模板。砂磨时间0 min的SiCNSs的TEM图如图5f所示,图中清晰可见所制备产物为薄片状,存在大小为5~10 nm的孔,即为多孔层状碳化硅。关于SiCNSs的生长机理将在后文讨论。

图5 (a~e)不同砂磨时间前驱体制备的SiCNSs的FESEM图;(f)砂磨时间0 min前驱体制备的SiCNSs的TEM图Fig.5 (a-e)FESEM images of SiCNSs prepared with the precursors sanded for different times;(f)TEM image of SiCNSs prepared with the precursor sanded for 0 min
2.3 吸附-脱附曲线与比表面积分析
图6为石墨薄片和SiCNSs的吸附-脱附曲线,可以看出,二者的吸附-脱附等温曲线均属于Ⅳ型等温线[15]。从图中还可以看出,与石墨薄片类似,SiCNSs等温线的回滞环同样属于H3型,进一步表明二者的结构具有相似性,即片状结构,这也与上述SEM结果一致。砂磨时间为0、5、30、125和400 min的石墨薄片作前驱体得到的SiCNSs比表面积分别为149、119、103、99、88 m2·g-1,即随着砂磨时间的增加,所得样品的比表面积呈减小趋势。与本课题组前期以高比表面积活性炭(2 161 m2·g-1)为碳源制得的3C-SiC纳米颗粒(84 m2·g-1)相比,二维的SiC纳米片状结构具有更高的比表面积。这意味着SiCNSs表面反应活性位点增多,更有利于提升其光解水产氢性能。
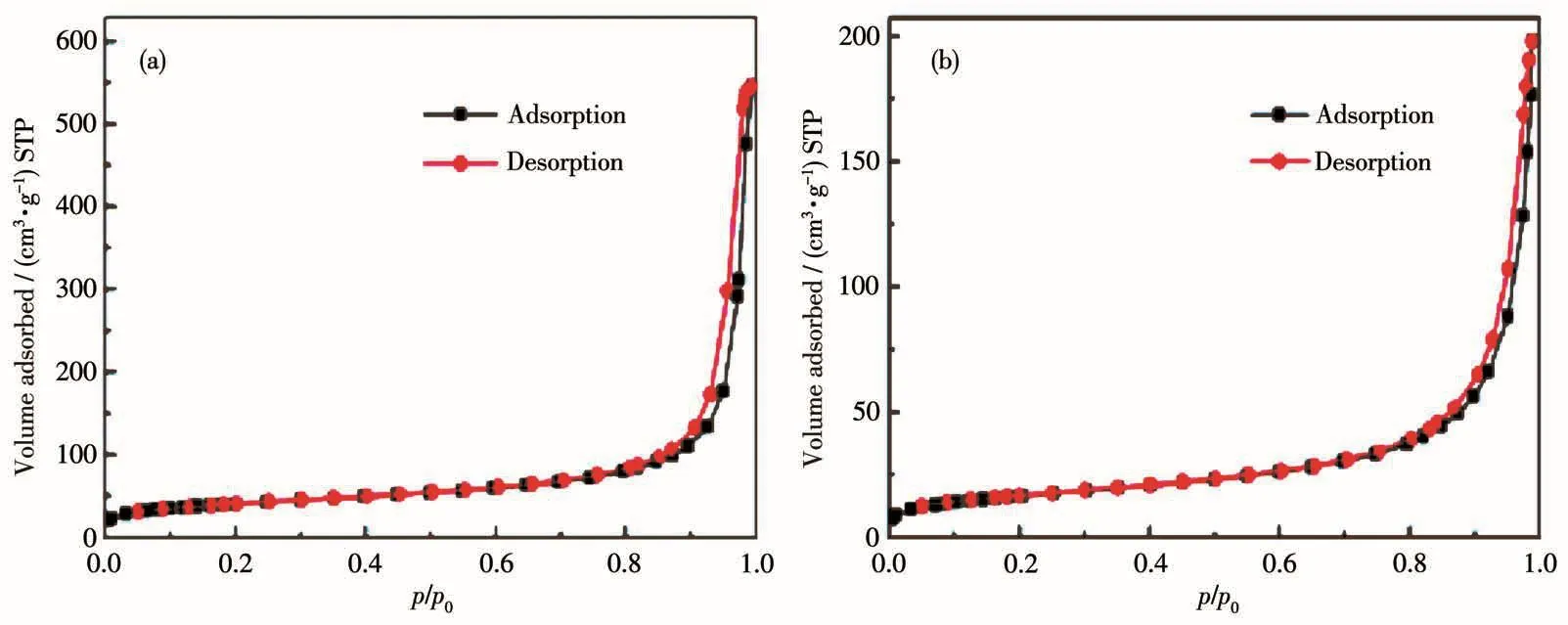
图6 石墨薄片(a)和SiCNSs(b)的吸附-脱附等温线Fig.6 Adsorption-desorption isotherms of graphite flakes(a)and SiCNSs(b)
2.4 紫外可见漫反射光谱(UV⁃Vis DRS)分析
通过UV-Vis DRS研究了样品的光学吸收性能。从SiCNSs的UV-Vis DRS谱图(图7)可以看出,样品在波长200~600 nm范围内有吸收,说明其具有较宽的可见光吸收区域,适合作为可见光照射下光解水产氢的半导体材料。样品的吸收边出现在550 nm左右,根据公式:Eg=1 240/λ,粗略计算得出禁带宽度Eg为2.25 eV,与3C-SiC的理论禁带宽度2.20 eV接近。
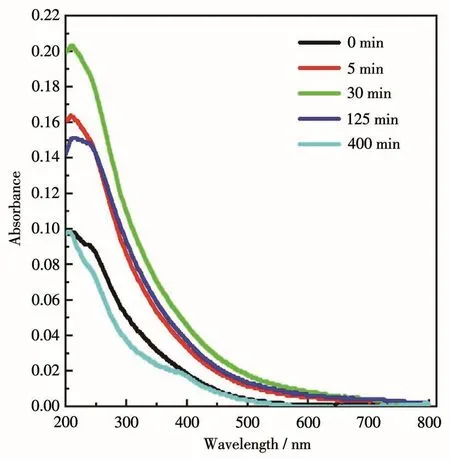
图7 不同砂磨时间前驱体制备的SiCNSs的UV-Vis DRS谱图Fig.7 UV-Vis DRS spectra of SiCNSs prepared with the precursors sanded for different times
2.5 石墨薄片制备SiCNSs的反应机理
本工作中SiCNSs是由石墨薄片作为碳源,高温反应制得。从前文的产物表征结果来看,石墨薄片的结构与形貌是制备SiCNSs的重要影响因素。图8是石墨薄片的XRD图,在2θ=26.6°处出现一个十分尖锐的衍射峰,其对应于石墨的(002)晶面,说明石墨薄片的片层排列较为有序,晶体结构完整。这表明石墨薄片依旧保持着石墨的晶体结构。图9a和9b是砂磨0 min石墨薄片放大不同倍数的FESEM图。从图9a中可以明显看到,石墨薄片呈片状结构,尺寸大小在30~50µm之间,周围存在脱落的小碎片;从图9b中可看到,石墨薄片表面较为平整、光滑,无较多褶皱。图9c和9d是砂磨400 min后石墨薄片放大不同倍数的FESEM图。经过机械砂磨后,石墨薄片平整的片状结构被破坏。从图9c中可看到,石墨薄片的尺寸在200~300 nm之间,且大小较为均匀。而图9d中,石墨薄片呈现团聚状态。这是由于纳米石墨片之间存在较强的范德华力作用。
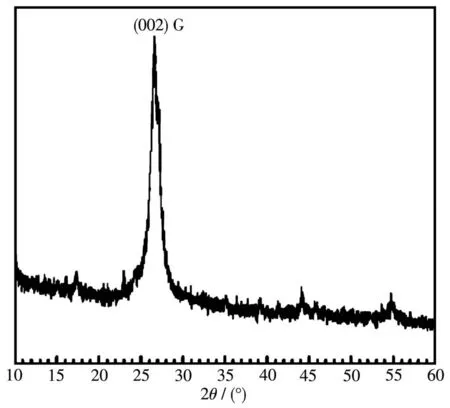
图8 石墨薄片的XRD图Fig.8 XRD pattern of graphite flakes

图9 (a、b)砂磨0 min和(c、d)砂磨400 min的石墨薄片的FESEM图Fig.9 FESEM images of graphite flakes sanded for(a,b)0 min and(c,d)400 min
目前对于SiC纳米材料的成核及其生长过程尚无一个明确的定论。关于其形成机制的解释主要有2种:一种是气液固(VLS)生长机制,另一种是气固(VS)生长机制。在我们所制备的SiCNSs的FESEM图中,均没有在纳米片周围发现符合VLS生长机制的小球。因此,可以认为SiCNSs的生长遵循VS机制。其可能出现的反应归结如下:
2C(s)+SiO(g)=SiC(s)+CO(g)
SiO(g)+3CO(g)=SiC(s)+2CO2(g)
C(s)+CO2(g)=2CO(g)
Si(g)+C(s)=SiC(s)
综合前文中对反应物和产物的结构、形貌等测试结果分析,我们提出SiCNSs形成机理如图10a所示。反应物经加热搅拌至糊状烘干,得到混合均匀的反应前驱体。当开始加热后,随着温度的上升,SiO(s)和 Si(s)蒸发气化,变成 SiO(g)和 Si(g)并与 C 发生反应。在石墨薄片表面形成众多的SiC晶核。在新形成的多个SiC晶核中,其(111)晶面随机取向,随着保温时间的延长,多个SiC晶核沿(111)晶面择优快速生长。由于SiO(s)和Si(s)位于石墨薄片表面,生成的SiO(g)和Si(g)气体还未扩散便与石墨薄片发生反应。即在石墨薄片表面原位生成SiCNSs,产物继承了石墨薄片的片状结构(图10b)。初始反应物中nC∶nSi=2.5∶2,即在初始产物中存在少量未反应的C,在650℃氧化气氛下焙烧除碳,即得到具有许多不规则的大小孔的SiCNSs(图10c),孔的多少很大程度上决定了产物的比表面积。
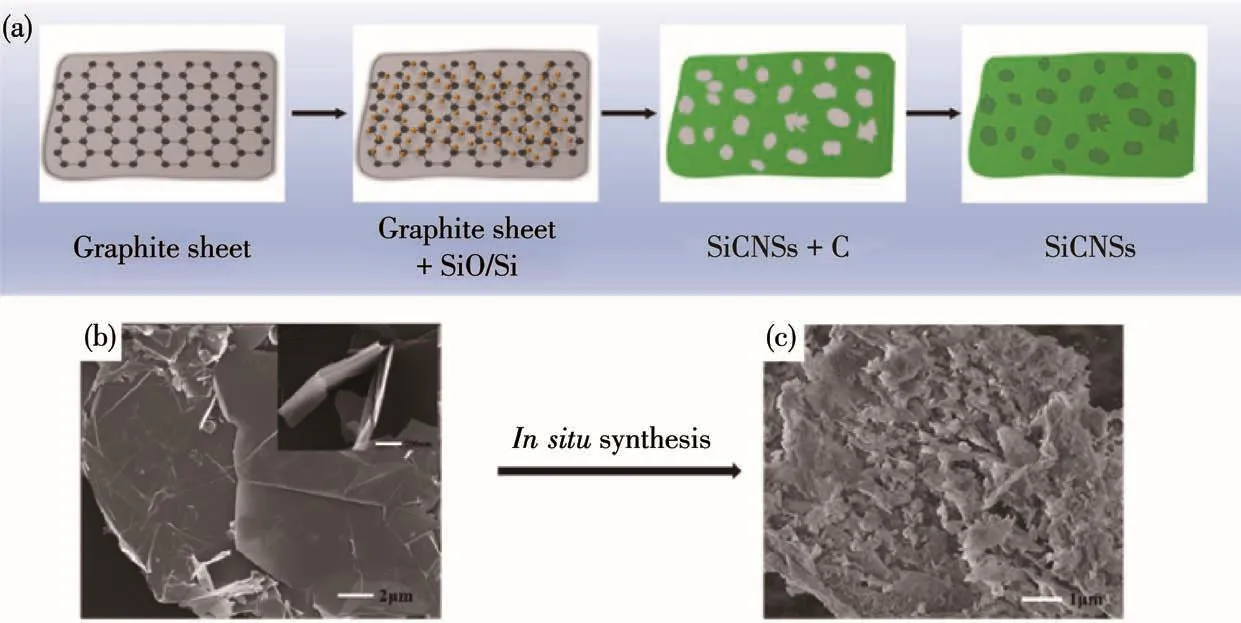
图10 (a)SiCNSs形成机理示意图;(b)石墨薄片的FESEM图;(c)SiCNSs的FESEM图Fig.10 (a)Schematic diagram of the formation mechanism of SiCNSs;(b)FESEM image of graphite flakes;(c)FESEM image of SiCNSs
图11为SiCNSs比表面变化示意图。高温下SiO和Si气化并与石墨薄片反应,首先在石墨薄片表面及四周形成SiC晶核。随着保温时间的延长,晶核长大。对于砂磨0 min的石墨薄片而言,由于初始反应物的粒径较大,比表面积较小,与SiO和Si的反应不能充分进行,这些众多的晶核难以在2 h内生长成完整的片状结构,因此有较多的C残留,经除碳处理后留下许多不规则大小的孔,这些孔的存在使得产物具有较高的比表面积。随着砂磨时间的增加,石墨薄片的单位尺寸逐渐减小,比表面积增大,同一反应条件下,反应物接触更完全,因此初始产物中C残留减少。对于砂磨400 min的石墨薄片而言,由于其粒径较小,高温下与气态的SiO和Si反应完全,形成的片状结构完整,这使得产物的比表面积较小。因此,基于上述形成机理,以砂磨0 min的石墨薄片作为碳源合成的SiCNSs,不仅继承了初始反应物的形貌,还具有较大的比表面积,这与比表面积测试结果一致。
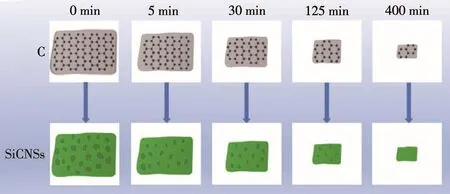
图11 SiCNSs比表面积变化示意图Fig.11 Schematic of the change in specific surface area of SiCNSs
2.6 SiC纳米片光解水产氢性能
在可见光照射条件下,比较了所制备材料作为半导体光催化剂在纯水中光催化产氢性能。图12a是不同比表面积的SiCNSs的产氢量随光照时间变化的曲线。从总体上看,所制备的样品随着光照时间的延长,其产氢量呈线性增加。图12b给出了不同比表面积的SiCNSs产氢速率。砂磨0、5、30、125、400 min石墨薄片制得的SiCNSs比表面积分别是 149、119、103、99、88 m2·g-1,对应产氢速率分别为51.0、36.0、16.8、14.4、9.2 µL·g-1·h-1。从以上数据可以看到,随着光催化剂比表面积的增大,其产氢性能显著提升。这主要是因为SiCNSs在水中分散后,受光源照射,激发产生大量的光生电子,这些光生电子迅速迁移至SiCNSs表面的活性位点,参与分解水制氢的还原反应。因此比表面积越大,SiCNSs表面的活性位点越多,越有利于光催化分解水反应的进行,从而使产氢性能得到提升。为了考察SiCNSs光催化剂的稳定性,对其进行了循环产氢测试。如图12c所示,在循环测试4次后,其产氢量并未随着时间的延长而明显下降,比较稳定。
3 结 论
利用几种廉价易得的工业原料(可膨胀石墨、SiO和Si粉),通过简单温和的方法制备了多孔的SiCNSs,并提出了SiCNSs的形成机理。此外,在上述材料制备的基础上,对SiCNSs进行了光催化产氢性能的研究。主要结论如下:
(1)通过机械砂磨得到不同粒径的石墨薄片,并以此为模板合成了不同比表面积的SiCNSs。
(2)通过对反应物和产物的结构和形貌对比分析,提出了以石墨薄片为模板原位生成SiCNSs的形成机理,该过程主要遵循气固反应机制。高温下,气态的SiO和Si与石墨薄片反应生成SiCNSs,产物较好地继承了石墨薄片的片状结构。
(3)SiCNSs的比表面积对其产氢性能影响显著,提高光催化剂的比表面积有利于增强其产氢活性。光解水产氢速率最高为51 µL·g-1·h-1。
猜你喜欢
疯狂英语·初中版(2022年3期)2022-03-31
小学生学习指导(高年级)(2022年3期)2022-03-29
有色金属科学与工程(2022年1期)2022-03-12
科普童话·学霸日记(2021年4期)2021-09-05
证券市场周刊(2020年46期)2020-12-28
小学生学习指导(高年级)(2019年4期)2019-11-27
牡丹江师范学院学报(自然科学版)(2019年2期)2019-09-10
学生天地·小学低年级版(2017年10期)2017-12-11
阅读与作文(小学低年级版)(2017年10期)2017-10-27
小学生导刊(高年级)(2017年2期)2017-06-10
