
Effects of viremia and CD4 recovery on gut “microbiome-immunity” axis in treatment-naïve HIV-1 -infected patients undergoing antiretroviral therapy
2022-03-16 06:22:20EddaRussoGiuliaNanniniGaetanaSterrantinoSebleTekleKirosVincenzoDiPilatoMarcoCoppiSimoneBaldiElenaNiccolaiFedericaRicciMatteoRamazzottiMarcoPallecchiFilippoLagiGianMariaRossoliniAlessandroBartoloniGianlucaBartolucciAmedeoAm
Edda Russo, Giulia Nannini, Gaetana Sterrantino, Seble Tekle Kiros, Vincenzo Di Pilato, Marco Coppi, SimoneBaldi, Elena Niccolai, Federica Ricci,Matteo Ramazzotti,Marco Pallecchi,Filippo Lagi, Gian Maria Rossolini,Alessandro Bartoloni,Gianluca Bartolucci,Amedeo Amedei
Abstract
Key Words: HIV; Antiretroviral therapy; Microbiome-immunity axis; Microbiota;Cytokines; Short chain fatty acid; Inflammation; Immunological responders; Viremia
INTRODUCTION
The mutual interaction between the human microbiota and the immune system defines the so-called “microbiome-immune axis”. This axis has also been associated with several diseases, including human immunodeficiency virus (HIV) infection[1 ].Indeed, a key place for HIV replication is the gastrointestinal tract. HIV replication in the gastrointestinal tract results in a severe depletion of CD4+T cells that leads to decreased function of the epithelial barrier, allowing microbes and microbial products to be translocated, which contributes to the chronic inflammatory response[2 ]. HIV replication can also result in a microbial dysbiosis condition[3 -5 ], which has been correlated with increases in markers of disease progression, immune activation, and microbial translocation[3 ,5 -7 ]. Notably, HIV-infected people harbour a distinct gut microbiota (GM)[8 ,9 ] with aPrevotella-rich community composition, typically observed in individuals from agrarian cultures or with carbohydrate-rich, protein- and fat-poor diets[10 ]. In addition, the significant subversion of theBacteroidetesandProteobacteriaphyla, with an imbalancedPrevotella/Bacteroidesspecies ratio and an abundance inEnterobacteriaceae,is one of the most persistent changes documented in untreated HIV infection[11 -13 ]. Moreover, the increased number of gut-resident bacteria capable of directly producing inflammation can be a probable mechanistic link between HIVassociated dysbiosis and high systemic immune activation[14 ]. However, converging data from cross-sectional studies suggest that the GM composition and its related immune response can change over the progression of HIV infection. In particular,correlating the composition of the gastrointestinal tract microbiome to immune activation, circulating bacterial products and clinical parameters, a decrease of commensal species, and a gain of pathogenic taxa was observed in HIV+ subjects compared to controls[15 ]. Additionally, analysing the functional gene content of the GM in HIV+ patients and the metabolic pathways of the bacterial community associated with immune dysfunction, the metagenome sequencing revealed an altered functional profile with significant interactions between the bacterial community, their altered metabolic pathways, and systemic markers of immune dysfunction[16 ].Furthermore, analysing the associations between the innate lymphoid cell (ILC)cytokines and measures of virologic, immunologic, and microbiome indices, it was observed that inflammatory ILCs contribute to gut mucosal inflammation and epithelial barrier breakdown, important features of HIV-1 mucosal pathogenesis[17 ].Despite growing evidence that the GM has a role in HIV pathogenesis[11 ,18 -20 ], the results were contrasting, with some studies suggesting an influence and others no HIV influence on microbial diversity[1 ,21 ] and composition[22 ,23 ]. However, many studies on the GM in HIV-infected patients are often carried out with a lack of adjustment for confounding factors, such as diet and use of drugs[24 ,25 ].
Currently, antiretroviral therapy (ART) has increased the life expectancy of HIVinfected patients, approximating it to that of the general population[26 ]. Interestingly,chronic inflammation and GM alterations persist in patients virologically suppressed by ART[27 ]. These data implicate that re-shaping the microbiota may be an adjuvant therapy in patients commencing successful ART[28 ]. On the other hand, suppressive ART appears to have a limited effect on the restoration of the GM[13 ,25 ,29 ,30 ].Although the gut microbial composition of ART-treated people differs from that of untreated people, the former also have a different microbial community structure compared to the HIV-uninfected population[31 ,32 ]. These findings raise the possibility that persistent gut dysbiosis may play a role in the development of residual clinical illness after ART.
Currently, the CD4 /CD8 ratio is considered one of the best-used markers of immune reconstitution. Notably, a low CD4 /CD8 ratio is associated with an increased risk of non-AIDS-related diseases[33 ]. Furthermore, the differences between the elements of the microbiome-immune axis between patients with normalized or nonnormalized CD4 /CD8 ratio during ART have not been elucidated so far[34 ,35 ];however, this question is recognized as a current research gap.
Moreover, with a better understanding of the microbiota-immune axis, it is now known that in addition to the intestinal flora itself, its metabolites are also involved in regulating vital host activities, such as energy metabolism, cell-to-cell communication,and host immunity. Short-chain fatty acids (SCFAs) are important metabolites able to modulate the production of immune mediators, such as key cytokines for the repair and maintenance of epithelium integrity[36 ]. In addition, the SCFAs modulate the activity of T cells and decrease the overexpression of histone deacetylase, particularly butyric and valeric acids[37 ]. SCFAs are an important link between microflora and the immune system; they involve different molecular mechanisms and cellular targets, are essential for the maintenance of intestinal homeostasis, and finally play a role in HIV infection[38 ].
The purpose of this prospective observational study was to compare for the first time the fecal microbial composition, serum and fecal microbial metabolites, and serum cytokine profile of treatment-naïve patients before starting ART and after reaching virological suppression (HIV RNA < 50 copies/mL) after 24 wk of ART. An additional aim was to correlate the GM composition, microbial metabolites, and cytokine profile of patients with CD4 /CD8 ratio < 1 and CD4 /CD8 > 1 after antiretroviral therapy.
MATERIALS AND METHODS
Patients
The study population, composed of 12 treatment-naïve HIV-infected patients receiving ART mainly based on integrase inhibitors, was enrolled between April 2018 and May 2019 at the Department of Infective and Tropical Disease at University Hospital of Careggi, Florence, Italy (Table 1 ). The study was approved by local institutional review boards and written informed consent was obtained from patients before participation (Rif CEAVC 15035 ).
We conducted a prospective observational cohort study comparing the changes occurring in the fecal microbiota, serum and fecal SCFA, serum free fatty acids (FFAs),and serum cytokines of patients with HIV-1 infection before ART (T0 ) and after 24 wk(T1 ). In addition, patients were divided into two groups according to whether they were immunological responders (IRs,n= 6 ) or not (INRs, n = 6 ) (INRs and IRs, based on the normalization of CD4 /CD8 ratio: < 1 or ≥ 1 after 24 wk of ART, respectively).Patients who had used antibiotics, probiotics, or prebiotics or had experienced diarrhoea or digestive symptoms within the previous 1 mo were excluded.
Personal data, ART regimen, HIV-RNA values, and number of CD4 + and CD8+T cells prior to ART starting and at the time of virologic suppression were included in the analysis (Table 1 ). In this pilot exploratory study, no formal sample size calculation was performed. All patients followed a Mediterranean diet.
Plasma HIV-RNA was measured using Test v1 .5 Roche COBAS AmpliPrep, Roche TaqMan HIV-1 Test v2 .0 (Roche Diagnostics, Branchburg, NJ, United States) and Siemens Versant K PCR (Siemens Healthcare GmbH, Erlangen, Germany), with lower limits of detection of 50 , 20 , and 37 copies/mL, respectively.
The T cell counts of patients were determined using a FACScanto flow cytometer(BD Immunocytometry Systems)[10 ]. Immunophenotyping of peripheral blood lymphocytes was analysed by three-color flow cytometry (Epics XL Flow Cytometry System; Beckman Coulter, United States) as previously described[39 ]. Freshly collected EDTA anticoagulated whole blood was incubated and tested with a panel of monoclonal antibodies directed against fluorescein isothiocyanate/phycoerythrin/peridinin chlorophyll protein combinations of CD3 /CD4 /CD8 , CD3 /CD16 CD56 /CD19 , HLA-DR/CD8 /CD38 , and CD4 /CD8 /CD28 and isotype controls (Immunotech,France).
At each time point (0 and 24 wk after study enrolment), we collected blood and fecal samples. After collection, stool samples were immediately frozen and stored at -80 °C until DNA extraction. Fecal samples were used to assess the microbiota composition and SCFAs, and while blood samples were used to measure SCFAs and FFAs and a panel of 27 selected cytokines.
Study follow-up
Patients underwent medical visits at 0 and 24 wk after study enrolment. They also underwent a comprehensive physical examination and medical history inquiry, urine toxicology panel testing, clinical laboratory tests including plasma HIV RNA,specimen collection, and detailed behavioural questionnaire survey. Demographic and clinical data were collected in a specific questionnaire and reported in an appropriate database, including the time point of follow-up in months; the participant’s gender,age, weight, and height; CD4 + and CD8 + T cell counts; the CD4 /CD8 ratio; HIV-1 RNA levels, ART, and antibiotic use. If subjects had to start antibiotics, they provided a last fecal sample and the study follow-up was immediately terminated.
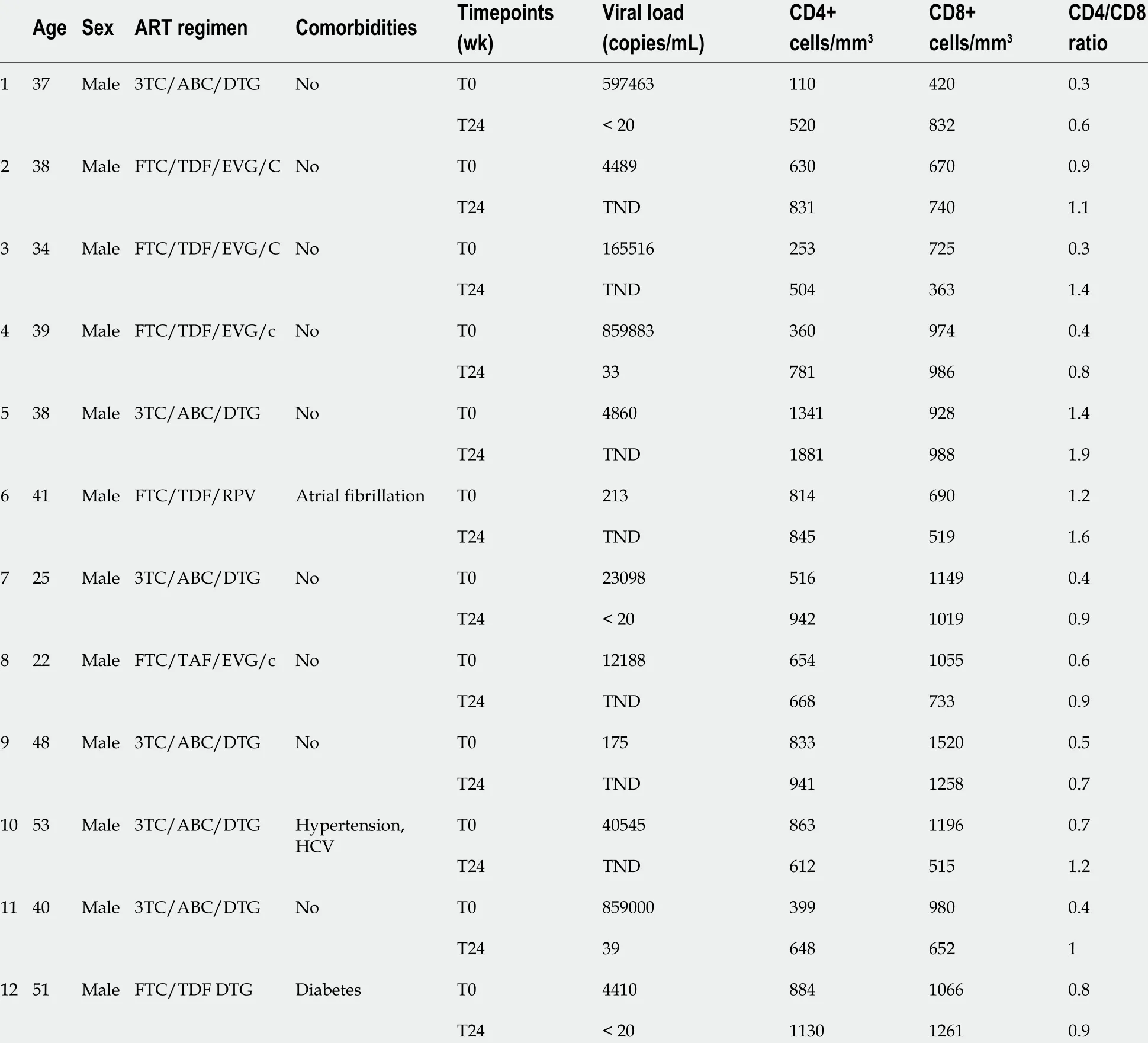
Table 1 Features of the enrolled patients
Fecal microbiota characterization
Total genomic DNA was extracted from frozen (-80 °C) stool samples, collected at different time points (weeks 0 and 24 ; T0 and T24 ), using the DNeasy PowerLyzer PowerSoil Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The quality and quantity of purified DNA were assessed using the NanoDrop ND-1000 (Thermo Fisher Scientific, WalthAP, US) and the Qubit Fluorometer (Thermo Fisher Scientific), respectively.
Extracted DNA samples were sent to IGA Technology Services (Udine, Italy) where amplicons of the variable V3 -V4 region of the bacterial 16 S rRNA gene were sequenced (2 × 300 bp paired-end) on the Illumina MiSeq platform, according to the Illumina 16 S Metagenomic Sequencing Library Preparation protocol[40 ].
Sequencing results were analysed using the QIIME 2 suite (Quantitative Insights Into Microbial Ecology)[41 ]. Briefly, following raw reads denoising (i.e., estimation of error rates, removal of chimeric and singleton sequences, and join of denoised pairedend reads) using DADA2 (Divisive Amplicon Denoising Algorithm 2 )[42 ], denoised reads were dereplicated and amplicon sequence variants (ASVs) were inferred.Taxonomic classification of inferred ASVs was performed using a Naive Bayes classifier trained on the SILVA 16 S reference database (release 132 ) (https://www.arbsilva.de/documentation/release-132 /).
Evaluation of fecal short chain fatty acids and serum free fatty acids by gas chromatography-mass spectrometry
The fecal SCFAs, in particular acetic, propionic, butyric, isobutyric, isovaleric, 2 -methylbutyric, valeric, and hexanoic acids, were analyzed using an Agilent GC-MS system composed with a 5971 single quadrupole mass spectrometer, a 5890 gaschromatograph, and a 7673 auto sampler. The chemicals, GC-MS conditions, and calibrations parameters are reported in supporting information (Tables S1 -S4 )[43 ].Fecal samples were collected in 15 -mL Falcon tubes and stored at -80 °C. Just before the analysis, each sample was thawed, weighted (between 0 .5 -1 .0 g), and added to sodium bicarbonate 10 mmol/L solution (1 :1 w/v) in a 1 .5 mL centrifuge tube. The obtained suspension was briefly stirred in a vortex apparatus, extracted in an ultrasonic bath (for 5 min), and then centrifuged at 5000 rpm (for 10 min). The supernatant was collected and transferred into a 1 .5 mL centrifuge tube (sample solution). The SCFAs were finally extracted as follows: An aliquot of 100 μL of sample solution was added to 50 μL of internal standard mixture, 1 mL of tert-butyl methyl ether, and 50 μL of 1 .0 mol/L HCl solution in a 1 .5 mL centrifuge tube. Afterwards,each tube was shaken in a vortex apparatus for 2 min and centrifuged at 10000 rpm for 5 min, and finally the solvent layer was transferred into an autosampler vial and analyzed by the GC-MS method. Each sample was prepared and processed, by the method described above, three times. In addition, serum FFAs, classified as SCFAs(acetic, propionic, butyric, isobutyric isovaleric, 2 -methylbutyri, and valeric acids),medium chain fatty acids (MCFAs; hexanoic, heptanoic, octanoic, nonanoic, decanoic,and dodecanoic acids), and long chain fatty acids (LCFAs; tetradecanoic,hexadecanoic, and octadecanoic acids) were analyzed with our previous described GC-MS protocol[44 ]. The chemicals, GC-MS conditions, GC-MS method, and calibrations parameters are reported in supporting information (Tables S5 -S7 ).
Just before the analysis, each sample was thawed. The FFAs were extracted as follows: An aliquot of 300 μL of plasma sample was added to 10 μL of internal standard mixture, 100 μL of tert-butyl methyl ether, and 20 μL of 6 M HCl plus 0 .5 mol/L NaCl solution in a 0 .5 mL centrifuge tube. Afterwards, each tube was stirred in vortex for 2 min and centrifuged at 10000 rpm for 5 min, and finally the solvent layer was transferred into a vial with a microvolume insert and analyzed.
Molecular inflammatory response in serum
The inflammatory response in serum samples of patients and healthy controls was evaluated using a specifically assembled kit ProCartaPlex MixMatch Human 27 Panel for Luminex MAGPIX detection system (Affymetrix, eBioscience) following the manufacturers' instructions.
In detail, the panel included macrophage inflammatory protein-1 α (MIP-1 α),interleukin (IL)-27 , IL-1 β, IL-2 , IL-4 , IL-5 , interferon gamma-induced protein 10 (IP-10 ),IL-6 , IL-8 , IL-10 , IL-12 p70 , IL-13 , IL-17 A, interferon (IFN)-γ, IFN-α, tumor necrosis factor-α (TNF-α), granulocyte-macrophage colony stimulating factor (GM-CSF),monocyte chemotactic protein 1 (MCP-1 ), IL-9 , P-selectin, IL-1 α, IL-23 , IL-18 , IL-21 ,soluble intercellular adhesion molecule-1 (sICAM-1 ), IL-22 , and E-selectin.
All measurements were performed in a blinded manner by a laboratory technician who was experienced in executing the technique. The levels of cytokines were estimated using a 5 -parameter polynomial curve (ProcartaPlex Analyst 1 .0 ).A value under the low limit of quantification (LLOQ) was considered as 0 pg/mL.
Statistical analysis
Statistical analyses on ASVs representing the bacterial community were performed in R (R Core Team, 2014 ) with the help of the packages phyloseq 1 .26 .1 [45 ] and DESeq21 .22 .2 [46 ], and other packages satisfying their dependencies, in particular vegan 2 .5 -5 [47 ]. Rarefaction analysis on ASVs was performed using the function rarecurve (step 50 reads), and further processed to highlight saturated samples (arbitrarily defined as saturated samples with a final slope in the rarefaction curve with an increment in ASV number per reads < 1 e-5 ). For the cluster analysis (complete clustering on euclidean distance) of the entire community, the OTU table was first normalized using the total ASV counts of each sample and then adjusted using square root transformation. The coverage was calculated by Good's estimator using the formula (1 - n/N) × 100 , wherenis the number of sequences found once in a sample (singletons), andNis the total number of sequences in that sample.
Richness, Shannon, Chao 1 , and evenness indices were used to estimate bacterial diversity in each sample using the function estimate_richness from phyloseq[45 ]. The evenness index was calculated using the formula E = S/Log(R), where S is the Shannon diversity index and R is the number of ASVs in the sample. Differences in all indices were tested using a paired Wilcoxon signed-rank test. The differential analysis of abundance at the ASVs as well as at the different taxonomic ranks (created using the tax_glom function in phyloseq) was performed with DESeq2 [46 ] using a two group blocked by patient design in order to perform a paired test[48 ].
In addition, the software GraphPad Prism (v. 5 ) and Statgraphics Centurion XVI software were used for immunological data analysis. Numerical data are presented as the mean ± SD. The concentrations of several cytokines in some of the samples lay below the curve fit of the standards. To avoid the bias that would have been introduced by excluding these data, the concentrations of the implicated cytokine were set at half of the lower cut off of the test system, which was usually about 1 pg/mL.Outliers at the other end of the spectrum (higher than the mean ± SD) were identifiedviaboxplots and were excluded from the statistical analysis. The comparisons between dependent groups were evaluated by the Wilcoxon matched pairs test, while the comparisons between the independent groups were assessed by the Mann-Whitney test. APvalue less than 0 .05 were considered statistically significant.
Data availability statement
The 16 S rRNA sequence dataset has been deposited in the NCBI Sequence Read Archive (SRA) database and is available under the BioProject accession number PRJNA731648 .
RESULTS
Comparison of fecal microbiota and metabolic and inflammatory profiles after ART
Modest differences in specific fecal microbiota taxa associated with HIV viremia:In the first part of our study, we compared the fecal microbiota and metabolic and inflammatory profile before and after ART starting, in order to examine potential changes resulting from HIV infection and ART therapy. We first analysed the longitudinal variation of fecal microbiota population in the same patients at T0 (HIV+viremia - RNA > 50 copies/mL), defined as “high viremia” condition, and T24 (HIV+suppression - RNA ≤ 50 copies/mL), defined as “viral suppression” condition. The alpha diversity of samples did not display significant differences for Chao, Shannon,and evenness indices (Figure 1 ). The analysis of the taxonomic composition revealed that more than 99 % of the sequences collected were classified into four phyla:Firmicutes(65 .46 %), Bacteroidetes (21 .54 %), Actinobacteria (9 .40 %), andProteobacteria(2 .72 %). In order to investigate similarity of patients’ microbiota abundance profiles and to study the paired nature of sampling (i.e., high viremia conditionvsviral suppression condition), a cluster analysis and PCoA on normalized ASV counts were performed.
The hierarchical clustering evidenced that microbiota was not sufficiently altered after treatment (24 wk) to break individual compositions apart, resulting in a perfect matching of the two time points from the same patient (Figure 2 A). This result was also confirmed by the PCoA (Figure 2 B), which showed a substantial proximity of each patient at T0 and T24 , indicating that, overall, the abundance profile of the single patient was not affected by the 24 -wk therapy.
On the other hand, the paired comparison of the abundance of single microbial ranks revealed some significant (adj.P< 0 .05 , abs (logFC) ≥ 1 ) differences between the two samples groups. In particular, the generaRuminococcus 2andSuccinivibriowere found to be significantly increased in higher viral suppression condition. On the contrary, viral suppression was related with a decrease in theIntestinibactergenus(median abundance, ~1 %) (Figure 3 ).
Analysis of fecal SCFAs displays no different layout between “high viremia” and“viral suppression” conditions:As we noticed minor changes in fecal microbiome profile (just at the order and genus levels), we wondered if the GM metabolic activity had been altered as well, and whether this activity might be masked by simply examining the microbiota composition. In order to evaluate the presence of alterations in GM metabolic activity, the levels of microbial linear and branched SCFAs were measured in fecal samples for each patient. However, the analysis of linear SCFA(acetic, propionic, butyric, and valeric acids), and branched SCFA (isobutyric,isovaleric, and 2 -metilbutyric acids) abundance did not reveal any significant change after 24 wk of therapy for each patient.

Figure 1 Box-plots showing alpha diversity indices (Chao1 , Shannon, and evenness indices) in samples. Statistical differences were evaluated using paired Wilcoxon signed-rank test for Chao, Shannon, and evenness indices. P value less than 0 .05 were considered statistically significant.
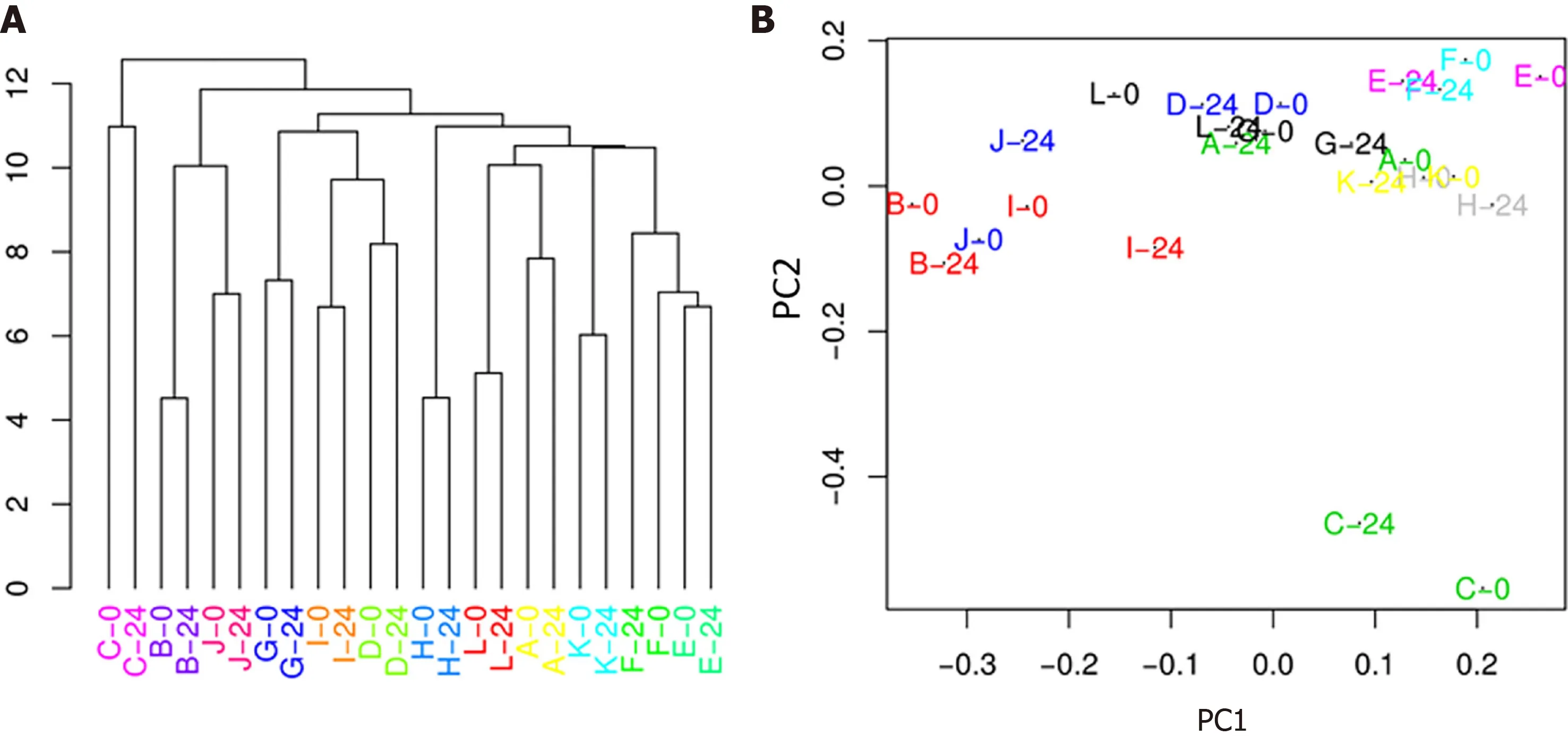
Figure 2 Cluster analysis (A) and principal coordinate analysis showing that samples do not separate into two groups depending on their condition (0 -24 wk) (B).
Analysis of serum FFAs reveals a significantly different subgroup of SCFAs between “high viremia” and “viral suppression” conditions:As we did not report alterations in the composition of fecal SCFAs, we wanted to observe if there were any other alterations in metabolic output, by analyzing both microbial and host derived FFAs in serum. As known, the impairment of gut integrity due to dysbiosis condition,leads to translocation of microbial elements from the intestinal mucosa to the bloodstream, which is considered a major driving force of chronic immune activation[49 ] even in patients successfully treated with ART and achieving stable virological suppression[2 ].
The analysis of serum FFA levels showed a significant change of two SCFAs at T24 compared to the baseline. In particular, propionic and butyric acids were increased in viral suppression condition (Figure 4 ).

Figure 3 Segment plots depicting taxa with significantly differences between high viremia (time point 0 ) and viral suppression (time point 24 ) conditions. Lines connect paired samples and highlight the differences in normalized abundance for the indicated rank. Orange or blue colors highlight decrease or increase, respectively. Numbers in the top-left corner represent counts of increased (orange) and decreased (blue) measurement for paired samples.
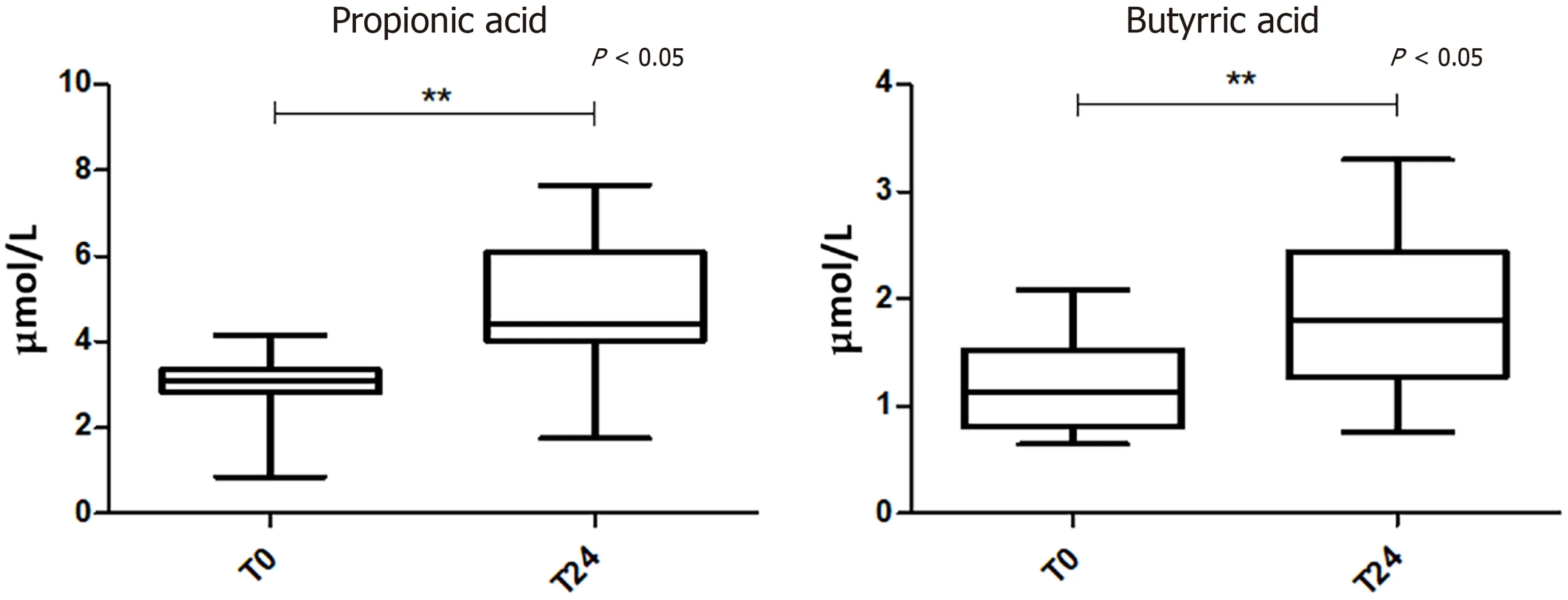
Figure 4 Boxplots showing statistically different levels of serum short-chain fatty acids between high viremia and viral suppressor patients, assessed by the Wilcoxon test. P value < 0 .05 was considered statistically significant.
Inflammatory profile between high viremia and viral suppression conditions:As known, gut microbial dysbiosis is linked to aberrant immune responses, as alterations in the GM may induce the interruption of gut epithelial barrier integrity with subsequent microbial translocation, increased inflammation, and immune activation,which are often accompanied by abnormal differentiation of immunological cells[6 ,50 ]. Since we detected significant variations of microbial communities between high viremia and viral suppression conditions, we decided to characterize also the serum immunological profile by evaluating a panel of 27 cytokines between the two mentioned conditions. Among the 27 cytokines examined, we detected a significant reduction of IP-10 (P = 0 .0244 ) and a significant increment of IL-8 levels (P = 0 .0547 ) in the high viremia setting (Figure 5 ).
Association of GM composition and metabolic and inflammatory profiles with CD4 +T-cell counts
Correlation between fecal microbiota and CD4/CD8ratio:In the second part of our study, we divided our cohort of patients into two groups: Immunological responders(IRs) and immunological non-responders (INRs), based on the CD4 /CD8 ratio > 1 or <1 . In this condition, the analysis of microbiota revealed that, considering only taxa with an overall abundance higher than 1 %, members of theFaecalibacteriagenus were significantly reduced (adj.P< 0 .05 , logFC = 1 .32 ) while members of theAlistipesgenus were significantly increased in responders (adj.P< 0 .05 , logFC = 2 .5 ) (Figure 6 ).
Different branched SCFA profiles in serum and fecal samples between IRs and INRs:As we observed significant variations in the composition of the fecal microbiota between IRs and INRs, we assessed if there were any other alterations in the fecal and serum microbial metabolites as linear and branched SCFAs derived from bacterial metabolism. We documented significant changes in isobutyric (P=0 .01 ), isovaleric (P=0 .04 ), and 2 -methylbutyric (P = 0 .04 ) acids, which were increased in IR fecal samples while we did not detect significant differences in serum samples (Figure 7 ).

Figure 5 Boxplots showing statistically different levels of serum cytokines between high viremia and viral suppressor patients, assessed by the Wilcoxon test. A P value < 0 .05 was considered statistically significant.
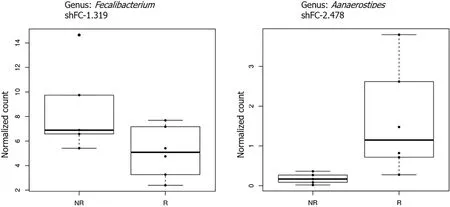
Figure 6 Boxplots showing the results of taxa-level differential abundance analysis between immunological responders and immunological non-responders at 24 wk. Plot titles report the shrunk Log2 fold change (according to the DESeq2 function lfcShrink). All results have a P value < 0 .05 . NR = INRs, R = IRs. IRs: Immunological responders; INRs: Immunological non-responders.
Inflammatory profile shows no significant differences between IRs and INRs:Since we detected significant variations of microbial communities between IRs and INRs, we also evaluated the serum immunological profile. However, cytokine levels did not show significant variations between the IRs and INRs.
DISCUSSION
Currently, the mechanisms regulating the interplay between the host immune system and HIV-1 , as well as the exact changes occurring in the GM composition and functionality, remain to be defined. To clarify the intricate relationships between the actors of the “microbiota-immunity” axis, we examined microbiota composition and functionality (SCFAs), serum inflammatory response, and FFA composition in individuals undergoing ART in different HIV infection settings.

Figure 7 Boxplots showing statistically different fecal short-chain fatty acid abundances between immunological responders and immunological non-responders, assessed by the Mann-Whitney test. aP value < 0 .05 was considered statistically significant.
Today, many studies on microbiota have been performed chiefly comparing HIVinfected and uninfected individuals, revealing a reduced GM diversity (the so-called HIV-associated dysbiosis) and an independent association between alpha-diversity of microbiota and peripheral levels of CD4+T cell count in treatment-naïve HIV-infected patients[28 ]. However, cross-sectional studies may not be suitable to provide information about cause-and-effect relationships, whereas longitudinal ones could be more valid for examining such relationships. Besides, there is a lack of human longitudinal observations of the “microbiota-immunity” axis before and after first ART administration. Only in few longitudinal studies, where HIV-1 -infected participants were followed after ART starting, data obtained on bacterial flora showed that shifts in the fecal microbiota persisted in a number of patients[10 ,28 ]. On the other hand, a recent study by Dillonet al[14 ] failed to find a significant change in a single time point study of the stool of HIV-1 -infected patients.
In this study, we first performed a longitudinal investigation evaluating the GM before the treatment and after “viral suppression” (T24 ). According to the longitudinal study conducted by Dillonet al[14 ], our results showed modest changes in the GM composition after ART; indeed, we did not assess significant differences in phylum composition. However, the paired comparison of the abundance of single bacterial taxa revealed a significant alteration at the genus level between the two sample groups(Figure 3 ). In particular, the genera ofRuminococcus,andSuccinivibriowere significantly increased after ART and the viral suppression. Conversely, the genus ofIntestinibacterwas significantly decreased in the same condition. We hypothesize that the slight change between the two groups may be due to persistent inflammation(related to microbial translocation and reduced immunoregulatory function), HIV latency throughout the gut, and direct effects of antiretroviral drugs on the bacterial population. Moreover, our results are in accordance with other longitudinal previous studies in non-human primates, which allowed to control for confounders affecting human studies[51 ,52 ]. We also reported an increase of the genusSuccinivibrio(Proteobacteriaphylum) between the two samples groups. In addition, in agreement with our data, the proportion of the rare genusSuccinivibrio, was also found considerably high in the stool of Japanese patients treated with ART[53 ]. One of the possible reasons for the contradictory results reported in the examined different studies may include the cross-sectional nature of the study, the used sampling method (stool swabvsstool),and the microbial taxon level applied.
Based on our findings, the 24 wk of ART inhibited HIV-1 viral replication effectively(indeed, all enrolled patients reached viral suppression), but did not heavily affect the overall bacterial composition of the gut microenvironment. The modest GM diversity that we observed between the two sample groups might be associated with the lowering of viremia. However, there was evidence that ART also induces changes in the gut microbiome, unrelated to HIV infection. Some authors have implied that ART may enhance dysbiosis, which is consistent with the high frequency of gastrointestinal side effects of this treatment[28 ,54 ].
As the GM influences the immune system through their bacterial metabolites, like SCFAs[55 ,56 ],we measured SCFA levels in blood and stool samples, in order to have a more accurate assessment of microbial metabolism after the ART. As known, the main SCFAs include, in order of proportion, acetic, propionic, and butyric acids that are produced by fibres fermentation by gut bacteria, particularly by members of theFirmicutesphylum[57 ]. Interestingly, for the first time, we observed a significant change of two serum SCFAs after the ART. In particular, propionic and butyric acids were increased in “viral suppression” condition. This altered SCFA profile may indicate a potential role for the SCFA synthesis pathway in the regulation of the HIV“microbiota-immunity” axis during effective ART. Notably, we did not observe any significant SCFA changes in stool samples, probably because in the colon, about 95 %of the produced SCFAs are rapidly absorbed by large intestinal mucosal cells while the remaining 5 % are secreted in the feces[58 ]. Propionate is only present at a low concentration in the periphery because it is metabolized in the liver[59 ]. It has been shown that butyrate may reduce gut inflammation by inducing the regulatory T cells (Tregs)and modulating activation of antigen-presenting cells[17 ]. We may speculate that bacterial flora responds reciprocally to inflammation by increasing the biosynthesis of anti-inflammatory and pro-solving lipid mediators that circulate in the bloodstream.Altogether, it is plausible that immune system-bacteria synergism mediates solutions to inflammation. On the contrary, as previously reported, some studies have found that butyrate-producing bacteria are selectively reduced in stool samples from HIVinfected compared to non-infected subjects[17 ,54 ]. In particular, Serrano-Villar et al[60 ]found that HIV-infected individuals had a distinct SCFA profile in stool compared to HIV-negative controls, with increased propionate and lower levels of acetate. No data from the literature are available regarding SCFA levels in HIV+serum samples, except a study of Segalet al[61 ] reporting that higher values of serum SCFAs, in consequence of an increased abundance of pulmonary anaerobic bacteria in HIV+ patients on ART,inhibited the immune response toM. tuberculosis, likely enhancing tuberculosis susceptibility. They observed that baseline serum butyrate and propionate were associated with the subsequent increasing hazard of tuberculosis. Moreover, we also evaluated serum FFA composition before and after ART treatment. Indeed, increased levels of FFA and proinflammatory cytokines have been reported in some HIVinfected patients under ART (reviewed in reference[62 ]). However, we did not appreciate any difference at the examined two time points.
Regarding the inflammation tone, there is consensus that a pro-inflammatory status remains active even after ART initiation in most patients[63 ,64 ]. Since the HIV life cycle is suppressed through ART in treated patients, the chronic inflammatory status observed in patients is maintained by factors secondary to HIV replication, including microbial translocation and reduced immunoregulatory function. In order to evaluate the inflammatory status after ART, we measured a panel of selected multifunctional effector molecules of the immune response in serum. Among the measured cytokines,we observed a decrease of IP-10 (P = 0 .0244 ) after the treatment, confirming the downregulation of this chemokine production in patients with HIV infection during ART[65 -69 ]. IP-10 is involved in trafficking immune cells to inflammatory sites, and it is considered an important pro-inflammatory factor in the HIV disease process. It has been observed that its levels can be reduced, but not to normal levels, by ART administration. Interestingly, IP-10 was consistently associated with HIV disease progression (based on CD4 + counts) during the period[70 ], suggesting its potential for use as an indicator of HIV infection and/or a therapeutic target for HIV treatment[71 ].On the other hand, in agreement with recent data, we observed a significant increased trend of IL-8 levels (P = 0 .0547 ) with suppressed viral load after 24 wk of ART. Indeed,increased IL-8 levels were observed in HIV-infected individuals on ART[72 ]. It has been shown that during HIV-1 infection, IL-8 plays an important role in the recruitment of CD4+T cells to the lymph nodes, thus generating more targets for viral replication. Our results may suggest that increased IL-8 Levels may represent a hallmark of chronic inflammation in HIV+patients on ART. In accordance with our findings, Wadaet al[73 ] observed significantly higher circulating IL-8 levels in HIV+men on ART with suppressed viral load in comparison to HIV-uninfected men.
It is now established that the gut microbiome may play a crucial role in the immune activation in HIV-infected patients treated with ART[5 ,64 ,73 -75 ]. Recently, several studies have reported that GM is associated with CD4+T cell recovery in HIV-infected patients, playing an essential role in the reconstitution of immune function[76 -78 ]. The potential mechanism includes the formation of a virus shelter, resistance to ART,promotion of intestinal mucosal barrier damage, and further entry of intestinal bacteria and their metabolites into the circulatory system, resulting in long-term immune activation, inflammation, and metabolic disorders such as cardiovascular diseases, diabetes mellitus, liver steatosis, and lastly, cancer[8 ]. Although it remains unclear whether an altered immunity after HIV infection drives dysbiosis orvice versa,the gut dysbiosis, immune dysfunction, epithelial damage, and microbial translocation are still evident even in the setting of ART-mediated viral suppression, which might be the treatment dilemma for HIV infection at present. Despite numerous studies of the microbiota in HIV-infected patients, there are relatively few reports discussing the compositional GM changes in patients with different immune responses to ART[79 ,80 ].
To investigate the role of GM in immunomodulation and immune reconstitution and which bacterial metabolites are implicated, in the second part of the study, we divided the patients into two groups: Patients with CD4 /CD4 ratio < 1 with insufficient reconstitution of CD4 + T cells despite achieving virological suppression after 24 wk of ART and those with CD4 /CD8 ≥ 1 who reached a robust reconstitution of CD4+T cells. We found that theAnaerostipesgenus was significantly augmented in IRs; on the contrary, theFaecalibacteriumgenus was significantly increased in INRs. Notably,Faecalibacteriumhas been reported as the anti-inflammatory commensal genus[81 ]. It has been positively correlated with the CD4 /CD8 ratio and anti-correlated with inflammation markers and LPS in a recent study in HIV-infected patients[82 ].
Regarding microbial metabolites, we detected a significant increase in fecal isobutyric, isovaleric, and 2 -methylbutyric acids in the IRs. However, we found that the changes associated with the IR group were not evident in the blood. Based on our results, we hypothesized that changes at the genus level in the gut ecosystem in HIVinfected patients undergoing ART might thus be both a consequence and a potential cause of the recovery of systemic immunity.
Our study had some limitations. First, a low number of patients were enrolled to investigate the elements of the microbiota-immunity axis and it cannot determine whether the altered GM contributed to or was caused by immune dysfunction. Second,only the effects of 24 -wk ART were observed in our study, and to establish a more meaningful connection between GM and microbial/immune parameters, future studies should investigate the GM alterations and the restoration of immune function after long-term effective ART. Finally, the microbiota of feces was a proxy for GM in this study, which was the only realistic sample for a non-invasive study. However,fecal microbiota may only represent the GM composition in the lumen rather than on the mucosal surfaces, which is an important distinction because the mucosa-associated microbiota potentially interacts with the gut-associated lymphoid tissue in HIV-1 -infected patients directly.
CONCLUSION
Our results provided an additional vision about the impact of HIV infection, ART, and immune recovery in the microbiota-immunity axis at the metabolism level, which are an indicator of the active processes occurring in the gastrointestinal tract. In summary,we demonstrated that patients infected by HIV-1 , after reaching virological suppression with ART, displayed a fecal microbiota with unchanged overall bacterial diversity except for few genera. Although 24 wk of treatment with ART was effective,the systemic inflammatory tone was not completely restored despite the anti-inflammatory serum butyrate increment. In addition, we confirmed the role of the GM in immune reconstitution, with the possible implication of bacterial metabolites;however, changes in the gut ecosystem in HIV+patients undergoing 24 wk of ART may thus be both a consequence and a potential cause of the recovery of systemic immunity.
Future larger-scale, long-term ART and longitudinal studies that include functional metagenomic and metabolomic approaches to identify the roles of the specific differential phylotypes are required to better define the relationship between microbiotaimmunity axis and HIV-1 infection and to provide new insights into the targeted treatment, improving the immune recovery and dampening inflammation.
ARTICLE HIGHLIGHTS


Research methods
The authors enrolled 12 treatment-naïve HIV-infected patients receiving ART. Fecal microbiota composition was assessed through next generation sequencing, and a comprehensive analysis of a broad spectrum of cytokines in blood was performed through a multiplex approach. In addition, serum free fatty acid (FFA) and fecal short chain fatty acid (SCFA) levels were measured through GC-MS.
Research results
The authors compared microbiota signatures, FFA levels, and cytokine profile before starting ART and after reaching virological suppression. Modest alterations were observed on microbiota composition; moreover, in the same condition, we also observed augmented levels of serum propionic and butyric acids. A reduction of serum IP-10 and an increase of IL-8 level were detected in the viral suppression condition. Thereafter, the same components were compared between immunological responders and non-responders. Concerning the microflora population, we detected a reduction ofFaecalibacteriumand an increase ofAlistipesin immunological nonresponders. Simultaneously, fecal isobutyric, isovaleric, and 2 -methylbutyric acids were also increased in immunological non-responders.
Research conclusions
The results provid an additional perspective about the impact of HIV infection, ART,and immune recovery on the “microbiome-immunity axis” at the metabolism level.These factors can act as indicators of the active processes occurring in the gastrointestinal tract.
Research perspectives
Future larger-scale, long-term ART and longitudinal studies that include functional metagenomic and metabolomic approaches to identify the roles of the specific differential phylotypes are required to better define the relationship between microbiotaimmunity axis and HIV-1 infection and to provide new insights into the targeted treatment, improving the immune recovery and dampening inflammation.
 World Journal of Gastroenterology2022年6期
World Journal of Gastroenterology2022年6期
- World Journal of Gastroenterology的其它文章
- Comments on validation of conventional non-invasive fibrosis scoring systems in patients with metabolic associated fatty liver disease
- COVID-19 , liver dysfunction and pathophysiology: A conceptual discussion
- Gallbladder Burkitt’s lymphoma mimicking gallbladder cancer: A case report
- Validation of the PAGE-B score to predict hepatocellular carcinoma risk in caucasian chronic hepatitis B patients on treatment
- Atrophic gastritis and gastric cancer tissue miRNome analysis reveals hsa-miR-129 -1 and hsa-miR-196 a as potential early diagnostic biomarkers
- Recent advances in the diagnostic evaluation of pancreatic cystic lesions