
隐孔菌倍半萜类成分抗肿瘤的作用机制:基于分子对接方法
2022-03-07 08:09:50周凌云赵梓童陈云艳
南方医科大学学报 2022年1期
隐孔菌[(Pk.)Hubbard]为多孔菌科隐孔菌属真菌,一般生长在针叶树的死木、倒木、腐朽木上。作为一种云南地区民间药用植物,常被用作治疗气管炎和哮喘的药物。其脂溶性部分主要单体化学成分为倍半萜类,该类成分体外具有细胞毒性,对肿瘤细胞的增殖有抑制作用,但具体靶点和作用机制无深入研究。抗肿瘤作用涉及通路和靶标较多,文献对于倍半萜类化合物抗肿瘤靶标有一些报道。Yang等认为Hirsutanol通过增加活性氧(ROS)的产生诱导肿瘤细胞发生凋亡;Chabranol 致低分化胃癌细胞发生凋亡的分子机制可能与凋亡通路中p53、Bcl-2、Caspase-3基因的异常转录相关;Parthenolide与抑制PI3K/Akt 信号通路有关;人参中倍半萜成分抗肿瘤作用可能与降低Bcl-2和VEGF表达及激活p38MAPK蛋白通道有关。由于各个通路中仍含有众多的作用蛋白,且多数仍未能合成得到,通过实验手段,验证化合物对于各靶点作用强弱不易进行,比较盲目,耗时费力。通过虚拟筛选则可以快速得到相互作用的基本信息,可以为药物机制研究和药物设计提供重要的参考。分子对接是利用作图软件将小分子(配体)放置于大分子靶标(受体)的结合区域,再通过计算参数,从而预测两者的结合能力和结合方式。通过结合能力强弱,可以初步推测分子可能的作用机制,并能快速准确地描述药物与潜在靶标间的相互作用方式,为衍生物的制备提供科学依据,借助虚拟筛选,可以缩短了药物研发周期,减少研发费用。目前,已有利用分子对接技术来研究分子作用机制及进行活性组分的筛选。本研究以前期分离得到的有活性的倍半萜骨架分子为基础,通过在线反向找靶,寻找空间上与此类分子较为匹配的抗肿瘤靶标。另外,考虑到常见抗肿瘤通路中有些是通过结构修饰(氧化、磷酸酯化或酯化)的方式实现竞争性拮抗,因此将文献报道过的倍半萜类抗肿瘤常见分子通路也列入考查范围。本研究利用分子对接的方式考查活性分子与选定靶点的结合程度,推测其抗肿瘤主要作用靶点和相互作用方式,并结合实验所得该类分子活性数值,分析不同分子骨架具有不同生物活性的原因,利用分子动力学模拟方式考查该类分子与蛋白靶点结合的稳定性。该研究能够为隐孔菌中倍半萜类成分作用机制的研究和衍生物的制备提供科学依据。
1 材料和方法
1.1 材料
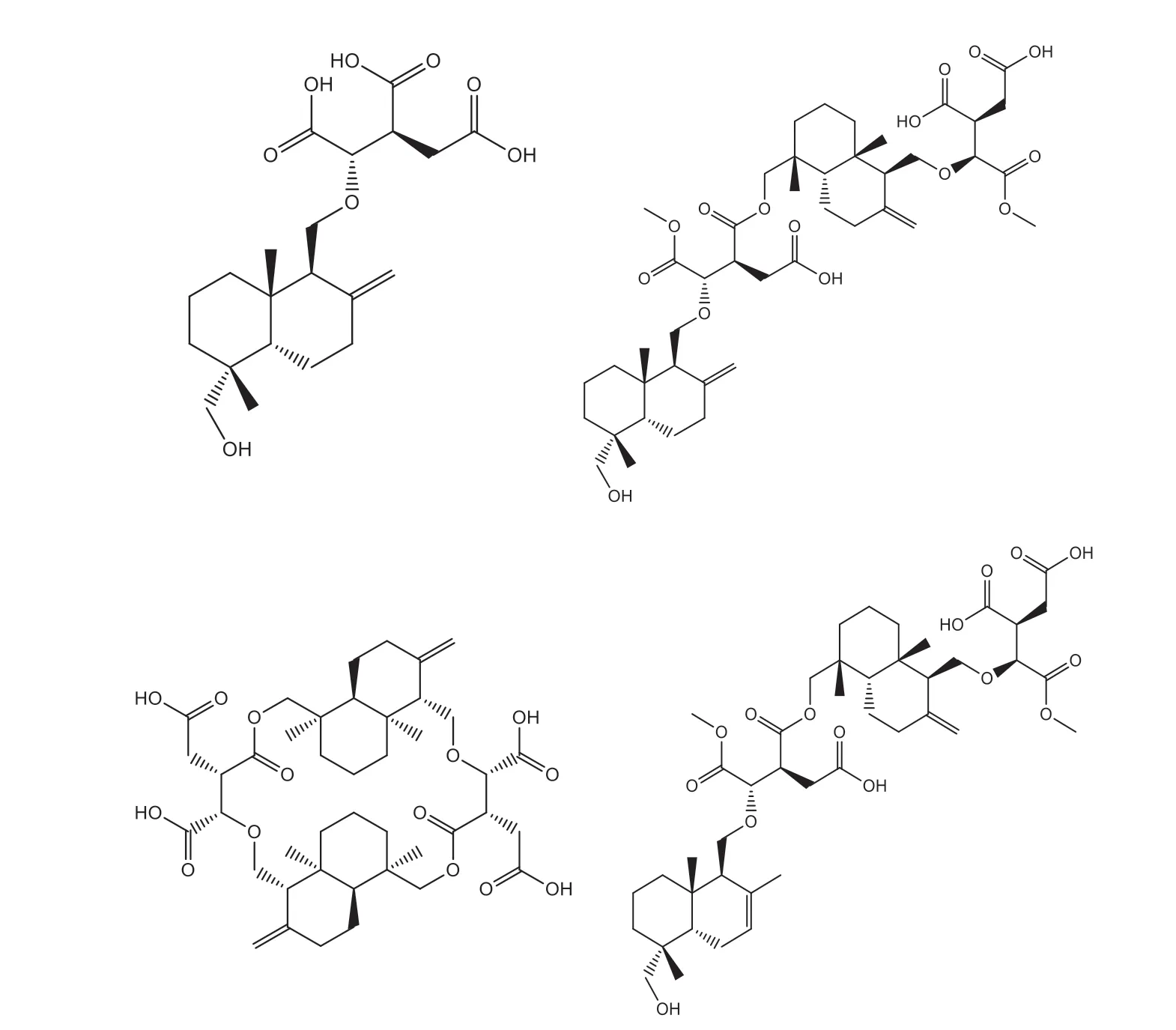
前期研究从隐孔菌子实体的乙酸乙酯提取物中分离得到19个倍半萜类化合物,它们分属四个骨架类型,选取各个骨架中体外细胞毒活性较高的分子作为代表,进行分子对接。它们的结构如图1所示,体外对五种肿瘤细胞的IC如表1所示。其中化合物4和化合物2体外细胞毒性较好,且对白血病HL-60细胞株毒性较强。
厦门某码头胸墙面层混凝土裂缝预防及控制措施…………………………………………………… 杨志文(10-189)
1.2 关键抗肿瘤靶点的选择及准备
抗肿瘤作用靶点众多,本研究从空间匹配程度及倍半萜类抗肿瘤常见特异性结合(如对靶标进行氧化、酯化等结构修饰)靶点两个方面入手进行初步筛选。通过在线投递化学结构,利用反向找靶网站PharmMapper(http://www.lilab-ecust.cn/pharmmapper)、SEA(http://sea.bkslab.org)、Target Hunter(https://www.cbligand.org/TargetHunter)进行初筛,这些网站均是由国内外不同高校开发,通过不同的算法,从不同的数据库(如PharmMapper 利用遗传算法,与来自TargetBank,DrugBank,BindingDB 和PDTD 这几个数据库,超过7000个受体基础的药效团模型进行匹配)中自动查询与小分子构象最为匹配的药效团,根据匹配程度进行排序,能够从空间匹配度入手迅速完成靶点的初步预测,从筛选结果中选出与抗肿瘤相关的靶点备用。同时,有些倍半萜类化合物能够与特定抗肿瘤靶点特异性结合,通过查阅文献,考查该类化合物常见抗肿瘤通路,结合蛋白质数据库PDB,从中选出清晰度高,结构解析清楚的人源靶标蛋白备用。从空间匹配和特异性结合两方面衡量,最终选择了14个靶蛋白作为分子对接的受体。
综上分析,船型尺度在考虑交通安全、过闸效率、应急救援、航道畅通等方面的现实条件影响下,无法针对所有船舶类型进行尺度放宽;同时,随着水运业供给侧结构性改革,对专业化、集约化船舶有更大体量的运输需要,服务于新兴业态发展有更深层次的诉求,因此尺度放宽界定为集装箱船、滚装船和江海直达船,意在先行先导,优势驱动。
在两个软件中分别选中蛋白分子和配体结合最优构象,DS利用“View Interactions”及“View Interactions(Advanced)”观察配体与蛋白分子3D 和2D 作用图。Maestro 中通过“Ligand Interaction”直接产生2D 作用图。
1.3 小分子配体的准备
国内PDA大致有两种形式,一是读者直接通过邮箱、论坛、到馆推荐的方式向图书馆提供所需文献的信息;二是图书馆与原版图书的出版商或是代理商合作,在图书馆举办大型原版书展,邀请读者参与,并根据他们的挑选情况进行现场采买。2014年5月,天津大学图书馆和中国进出口总公司就曾联合举办了这样的原版书展,参展的都是最新出版的原版外文图书,是经有关专家和国外出版社推荐,从剑桥大学出版社等众多国际知名出版社中精选出来的,他们让图书馆的读者现场参与挑选,最后,根据推荐情况进行择优订购。像这样让读者充分参与图书采购决策,能够增加原版外文图书的利用率,将滞架率降低。
1.4 分子对接
采用DS和Maestro软件进行对接。DS中靶点活性口袋的确定通过选中原晶体复合物中配体小分子,优先利用“From Current selection”或者直接选用“From PDB Site Records”,定义活性口袋,半径选择为10Å。Maestro 中活性口袋定义方法参照DS 中活性位点三维坐标确定。DS 中利用LibDock 工具,规定构象数量,通过“High Quality”对接,DS 中对接结果以“LibDockScore”来衡量不同分子及不同构象与蛋白受体对接情况。Maestro中通过XP(extra precision)进行“Ligand Docking”,对接结果以“Docking Score”来衡量。记录各个分子最优结合构象的得分情况。分子对接领域任何一种单独的策略并不具备绝对优势,不同的靶标需要利用不同的搜索算法和打分函数的组合来获得最优结果。本研究两个打分软件采用不同算法。Maestro运算时间稍长,精度稍高,Maestro Score得分越低说明结合越好;而Libdock 运算速度较快,Libdock Score得分越高,说明结合越好。
1.5 相互作用结果分析
靶蛋白三维结构从PDB(http://www.rcsb.org)中搜索下载,优先选择人源蛋白且分辨率较高的晶体结构,确定PDB 代码分别为5XP3(T2R-TTL)、2VPB(Bcl9)、2O6L(UDP-glucuronosyltransferase 2B7)、3HR4(iNOS)、1ORD(Inducible ornithine decarboxylase)、3BKY(β-lymphocyte antigen CD20)、3CKI(Metalloproteinase inhibitor 3)、1O4N(Proto-oncogene tyrosine-protein kinase Src)、3IW4(Protein kinase C-α)、2IOE(Protein kinase C-β)、1UNQ(Akt,序列1-123)、3QKL(Akt,序列144-480)、4QTB(Mitogen-activated protein kinase 3,MAPK3)、3HHM(Phosphatidylinositol 4,5-bisphosphate 3-kinase,PI3K),它们被用作为分子对接研究的载体。分别利用Discovery Studio 4.0(DS)和Maestro 12.3(Maestro)软件对蛋白结构进行补全不完整残基、删去配体分子和水分子、加氢和赋电荷等优化处理。
1.6 分子动力学模拟
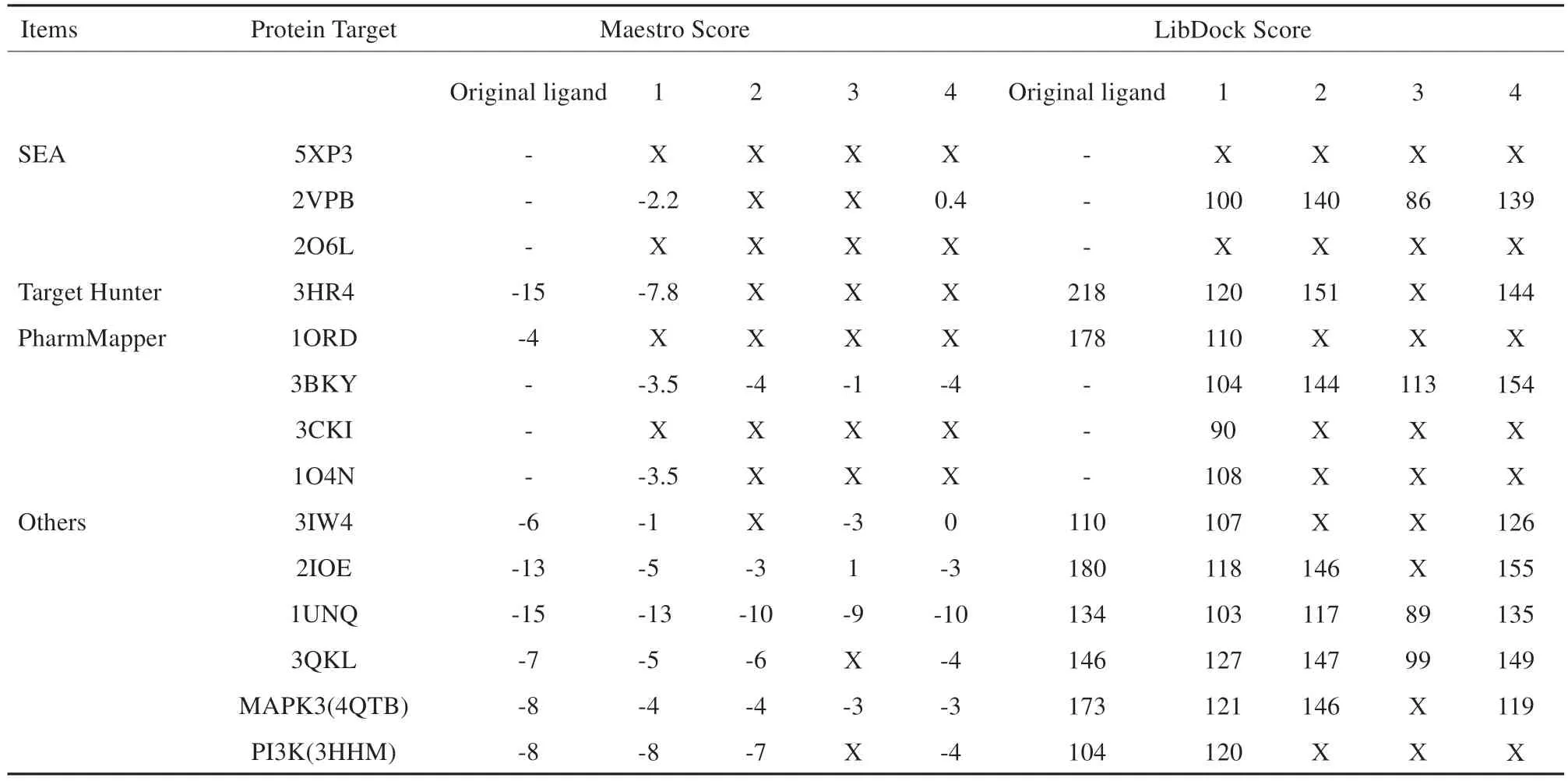
运用DS(Libdock)和Maestro对4个倍半萜类化合物和筛选出的14个蛋白序列进行对接打分,结果见表2。从结果可见,两个软件打分大体趋势是相同的,均显示各小分子配体与蛋白序列1UNQ有较高的对接打分,故认为该类倍半萜类化合物更容易与1UNQ匹配结合,推测1UNQ蛋白序列为该类物质主要作用靶点。
2 结果
2.1 四个倍半萜分子与潜在靶点的分子对接结果
通过DS软件分别对纯蛋白受体和对接得到的复合物进行分子动力学模拟。通过“Simulation”对制备过的各个分子结构增加CHARMM力场,利用“Solvation”溶剂化蛋白。对添加了溶剂体系的蛋白利用“Standard Dynamics Cascade”进行分子动力学模拟参数设置。升温时间设置为40 ps,平衡时间设置为400 ps,模拟采样时间设置为10 000 ps,模拟步长为2 fs,其他参数设为默认值。10 ns模拟结束后分析RMSD值和RMSF值。
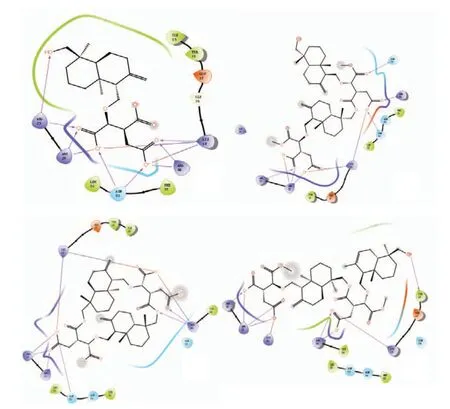
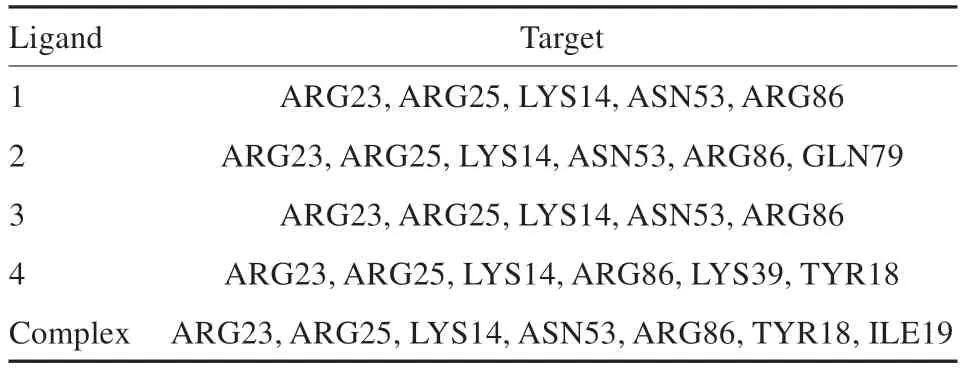
由Maestro 软件得到的4 个倍半萜类化合物与1UNQ最优结合的2D相互作用(图2),显示该类成分与受体的相互作用主要依靠羧基与蛋白之间以氢键和静电力相互作用。分子中的氧原子(红色部分)是其与蛋白靶点产生作用的主要基团。各化合物及蛋白复合物原配体与蛋白结合位点类似(表3),说明该类化合物与该活性口袋有良好作用。
2.2 四个倍半萜分子与蛋白序列1UNQ的2D相互作用图
你是左撇子吗?如果答案肯定,你就属于在人群中只占十分之一的少数派。在学习、生活中,也许你曾遭遇过一些尴尬和痛苦,比如,吃饭的时候怕和你的筷子打架,谁都不愿意挨着你坐。随着社会的开明程度不断提高,左撇子们的境遇变得越来越好,甚至在某些领域还独占鳌头。比如,在体育竞赛中,左撇子的击剑手、棒球投球手、拳击手,相比于同实力右撇子选手,则更容易获得胜利。再来看一个事实,在美国的倒数前7任总统中,有4位都是左撇子——福特、老布什、克林顿和奥巴马。左撇子并非生理缺陷,而是正常的遗传现象,科学家称它为“偏侧优势”。
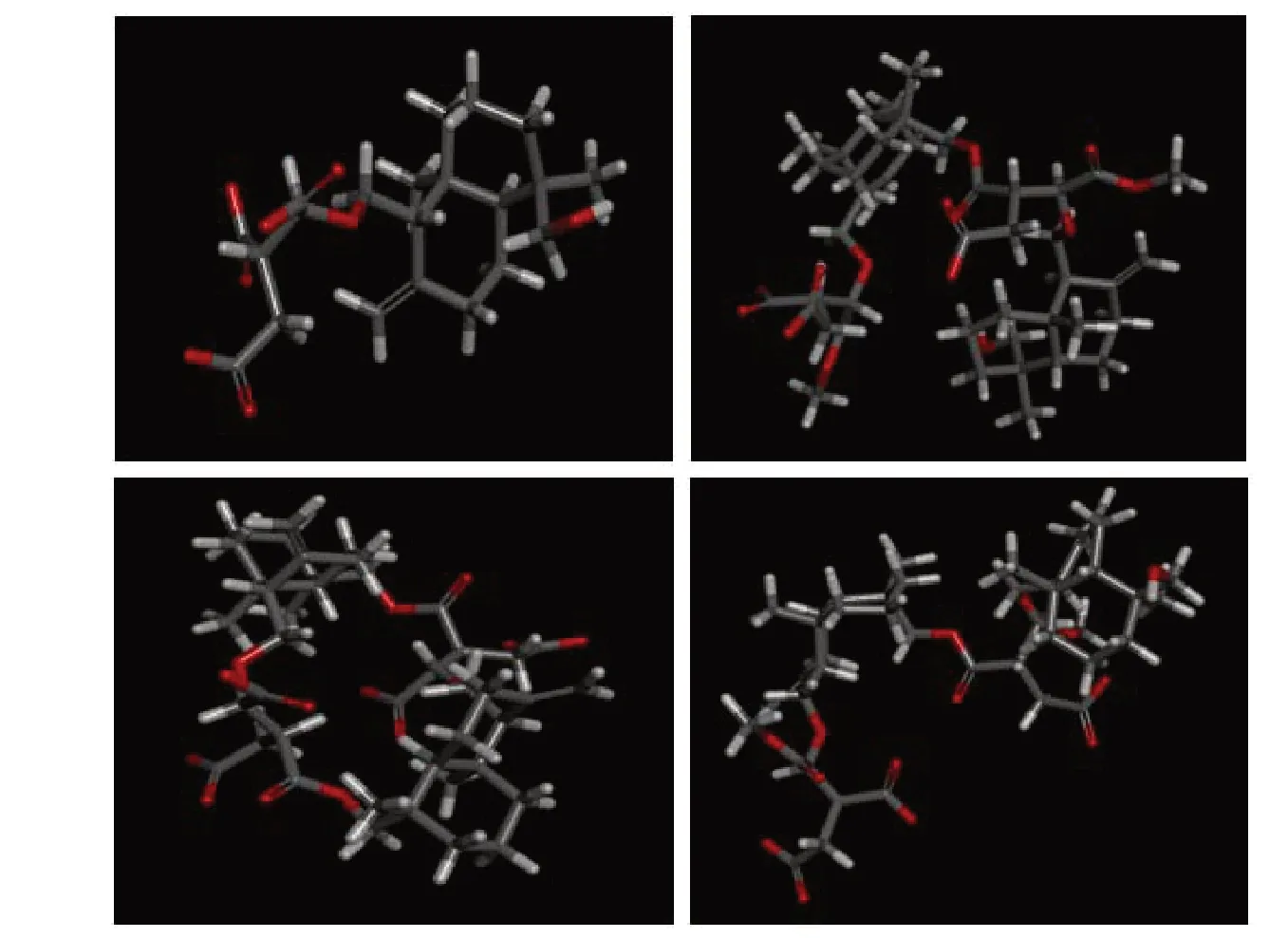
2.3 四个倍半萜化合物与1UNQ最优结合构象
本次研究采用的SiO2温标为无蒸汽损失SiO2温标、最大蒸汽损失在100 ℃SiO2温标、无蒸汽损失玉髓温标,阳离子温标主要采用Na-K、Na-K-Ca、K-Mg温标。
分子对接结果中四个倍半萜类化合物与蛋白靶点结合打分最高的构象(图3)即为该分子与1UNQ最优结合构象,可以发现四个不同的倍半萜化合物与靶蛋白结合的最优构象存在较大的差别。
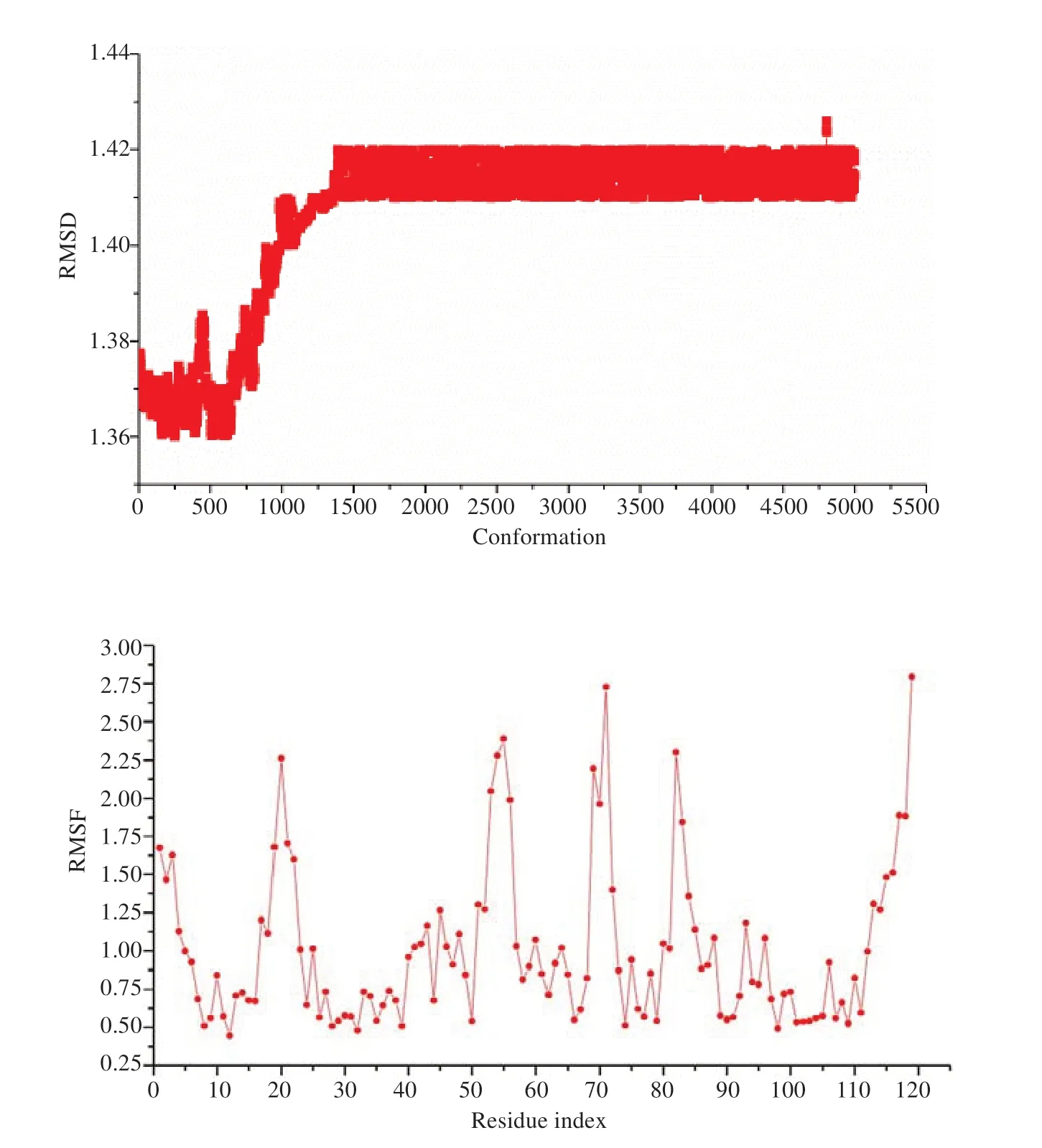
2.4 分子动力学模拟结果
比较1UNQ纯蛋白序列和化合物4结合后复合体结构的RMSD值,复合物RMSD值波动图见图4,其均值为(1.41±0.02)Å。约3 ns后整个复合物趋于稳定,无较大波动,相对于1UNQ纯蛋白波动值为(1.75±0.21)Å略低。复合物RMSF值较高的氨基酸序列为TYR18,ASN53,ARG69,ARP80。
以隐孔菌中分离得到的具有一定体外细胞毒性的4个化合物作为小分子配体。通过Chem 3D软件将化合物转化为三维结构,将其在DS分子窗口中复制粘贴到一个窗口中,并保存为mol2格式文件,对接前分别载入DS和Maestro中,通过赋予CHARMM力场,规定相应PH 范围等进行优化操作,用于与蛋白结构的分子对接。
3 讨论
本研究显示,四个倍半萜化合物体外对五个肿瘤细胞的体外细胞毒性强弱为化合物4>化合物2>化合物1>化合物3。不同的倍半萜骨架体外细胞毒性存在明显差异。通过反向找靶和分子对接技术,我们推测1UNQ蛋白序列为该类物质主要作用靶点。根据不同三萜骨架与1UNQ的2D相互作用图,不同化合物分子主要依靠羧基、羟基与蛋白靶点相应位点产生氢键或静电作用。体外细胞毒性最高的化合物4与蛋白靶点之间作用键最多且作用距离较短,结合更为牢固。另外,结合表3所列与配体结合具体作用靶标氨基酸,化合物4的C-15位较为特殊,此处的羟基能够与受体TYR-18位点产生氢键作用,推测该位置对于活性有较大贡献。
同时,根据四个倍半萜化合物与1UNQ最优结合构象进一步分析不同骨架对活性的影响,认为空间构象上的变化通过影响配体与靶标相互之间键合来影响其生物活性。化合物2由于双键在环内,使得双环处在离受体较近的空间位置,从而导致了分子与受体结合时产生了空间位阻,导致部分红色位点无法与相应的蛋白位点产生作用,降低了生物活性;化合物3由于形成了环合,其结构中的一个双环被固定在位阻位置,且为刚性结构,极大影响了分子与蛋白的结合,使得活性大为减小;化合物4中双键位于环外,通过自由转动使其结合最优构象中双环并不影响与蛋白靶点的结合,同时,C-15位羟基刚好与蛋白序列上TYR18形成氢键,增加了亲和力;化合物1(红色部分少一些)与1UNQ形成的相互作用力相对化合物2和化合物4较少,且由于分子较小,与相应靶点空间匹配度不如二聚体,结合较为松散。
通过分子动力学模拟对该类倍半萜化合物与1UNQ的结合稳定性进行验证,该技术能够模拟自然状态下两者的结合,可以还原一些目前的技术手段所不能检测到的反应过程,通过分析分子三维构象的变化,说明配体与受体结合的稳定性。化合物4与1UNQ复合体的RMSD值小于1UNQ的RMSD值,提示分子与1UNQ 有较稳定的结合。RMSF 值较高的位置则是1UNQ与化合物分子产生相互作用的邻近基团,这些位点构象变化较大,以利于更好与配体化合物产生结合,也使得结合形成的复合物较为稳定。
Akt激酶是一种重要的蛋白激酶,是细胞内重要的调控蛋白,Akt的活性与其自身的磷化、去磷酸化密切相关,且通过磷酸化下游底物蛋白和转录因子来影响细胞增殖的过程。本研究选择了Akt蛋白激酶中两段不同的序列1UNQ(1-123)和3QKL(144-480)进行研究,文献报道其行使磷酸化主要的靶点为Thr-308、Ser-473和Tyr-474,均存在于后端序列3QKL中。前端序列1UNQ中重要的靶点为Lys-14或Lys-20,这两个位置的乙酰化能够诱导Akt三级结构的破坏,使其无法与下游PIP(3,4,5-三磷酸磷脂酰肌醇)结合,从而阻断信号传导,进而对肿瘤细胞增殖起到抑制作用。对接的结果显示隐孔菌中倍半萜化合物与1UNQ作用比3QKL更好,说明该类化合物并不是通过占据后端蛋白作用靶点而起竞争抑制作用,更多是与前端蛋白序列有良好作用。从2D相互作用图可见,它们与Lys-14有相互作用,推测该类倍半萜类化合物能够引起Lys-14位乙酰化,导致Akt无法与下游PIP结合,影响细胞的增殖。
本研究选取体外实验有较好细胞毒性的隐孔菌倍半萜分子作为研究对象,通过反向找靶和文献调研初筛了14个可能作用靶点,利用分子对接技术,对倍半萜分子和可能靶点结合情况进行打分,最后确定蛋白激酶B的前端蛋白序列1UNQ为潜在的作用靶点。结合实际体外细胞毒数据,认为不同倍半萜骨架分子与靶蛋白结合存在不同的位阻,进而导致不同的生物活性。该类化合物与靶点有较稳定的结合,通过引起Lys-14位乙酰化,导致Akt无法与下游PIP结合,影响细胞的增殖。该研究对于进一步研究该类分子抗肿瘤作用机制及其衍生物的开发提供了科学依据。
