
POE-g-GMA对PLA/PP共混体系结构及性能的影响
2022-03-03 03:20:22谢永健陈鑫亮樊炳宇
石油化工 2022年1期
高 尚,谢永健,陈鑫亮,杨 利,樊炳宇,王 平
(安徽建筑大学 材料与化学工程学院 安徽省先进建筑材料国际联合研究中心,安徽 合肥 230601)
聚乳酸(PLA)因具有优异的机械性能和生物可降解性能,以及能用作石油基聚合物替代品[1-4]等特点,受到广泛的关注,但由于制品较脆等因素,限制了PLA基材料的广泛应用。通过共混改性是提高PLA综合性能的方法之一[5-8]。聚丙烯(PP)具有优良的综合性能,将其与PLA共混能够提高PLA的综合性能。但PLA与PP较弱的热力学相容性导致PLA/PP共混材料的性能较差。目前,添加交联剂或相容剂改善两相之间的界面状态或界面结构是提高PLA与PP相容性的主要方法之一,如苯乙烯-(乙烯-丁烯)-苯乙烯嵌段共聚物、乙烯-丙烯酸甲酯-甲基丙烯酸缩水甘油酯无规三元共聚物、过氧化二异丙苯、聚二甲基硅氧烷-聚乙二醇弹性粒子等[9-12]。Zawawi等[13]利用PP接枝马来酸酐以改善不同相之间的界面状态,并加入蒙脱石有机纳米黏土增强材料的机械性能。Savas等[14]利用乙烯-丙烯酸丁酯及碳纤维增强PLA/PP体系的相容性和机械性能,制备了强度更高的可再生复合材料。关于制备具有良好刚韧平衡性的PLA/PP共混复合材料的研究较多,但对PLA/PP共混复合体系界面形貌演变的过程和机理的研究报道较少。
本工作采用甲基丙烯酸缩水甘油酯(GMA)接枝乙烯-辛烯共聚物(POE)得到的POE-g-GMA作为相容剂引入到PLA/PP共混体系中,利用FTIR,DSC,SEM等方法研究了POE-g-GMA对PLA/PP共混体系加工性能、流变性能、结晶性能和机械性能的影响,并对POE-g-GMA的增容机理进行了探讨。
1 实验部分
1.1 实验原料
PLA:牌号4032D,Nature Works公司,Mw=195 000;PP:牌号JH380,Lotte Chemical公司,Mw=220 000;POE-g-GMA:牌号SOG-02,佳易容聚合物有限公司,GMA含量约2%(w)。
1.2 实验仪器
RM-200C型转矩流变仪:哈尔滨哈普电气技术有限责任公司;XH-406C型抽真空平板硫化仪:东莞市锡华检测仪器有限公司;WZY-4030型智能数控万能制样机:承德金和仪器制造有限公司;CMT4304型微机控制电子万能试验机:美特斯工业系统有限公司;Nicolet6700型傅里叶变换红外光谱仪:赛默飞世尔公司;DSC 30型差示扫描量热仪:上海精科天美贸易有限公司;DHR-2型旋转流变仪:TA公司;JSM-6490LV型钨灯丝扫描电子显微镜:日本电子制造株式会社。
1.3 试样制备
首先将所有原料在真空干燥箱中60 ℃下干燥24 h,然后按表1中的比例加入到转矩流变仪中,在180 ℃、转速50 r/mim条件下,熔融共混6 min。将所得材料充分冷却干燥后置于真空平板硫化仪中在180 ℃,10 MPa下,热压5 min再冷压3 min成型,并参考文献[15]的标准将材料裁成哑铃形样条。

表1 PLA/POE-g-GMA/PP共混物的组成与配比Table 1 Composition of PLA/POE-g-GMA/PP blends
1.4 测试与表征
采用傅立叶变换红外光谱仪研究POE-g-GMA对PLA/PP共混体系分子间相互作用的影响:在衰减全反射模式下,扫描波长范围为600~4 000 cm-1,扫描分辨率为4 cm-1,扫描次数为32次。
采用旋转流变仪表征试样的流变行为:通过万能制样机将试样制成直径为25 mm,厚度为0.8 mm的圆片,然后将圆片置于氮气流量为5 L/min的平行板旋转流变仪的平行板夹具中,然后升至180 ℃将试样熔融,恒温5 min消除热历史,并进行动态频率扫描,动态扫描条件为:固定应变1%,扫描角频率范围0.04~400 rad/s,进行小振幅振荡剪切,测试动态储能模量(G')、动态损耗模量(G'')和复数黏度(η*)随扫描角频率的变化关系。
采用差示扫描量热仪研究POE-g-GMA对PLA/PP共混体系凝聚态结构的影响:取10 mg左右试样,在氮气气氛下,以10 ℃/min升温速率升至200 ℃,并在200 ℃下等温5 min以消除热历史,然后以10 ℃/min的降温速率降至-30 ℃,最终以10 ℃/min的升温速率升至200 ℃进行二次升温。
采用微机控制电子万能试验机研究POE-g-GMA对PLA/PP共混体系机械性能的影响,通过万能制样机制成哑铃形样条,并以5 mm/min的拉伸速率进行拉伸测试。
采用扫描电子显微镜在加速电压5 kV下,观察PLA/PP/POE-g-GMA共混试样的拉伸断面。
2 结果与讨论
2.1 POE-g-GMA对PLA/PP共混体系加工性能的影响
图1为PLA/POE-g-GMA/PP共混体系的扭矩-时间曲线。
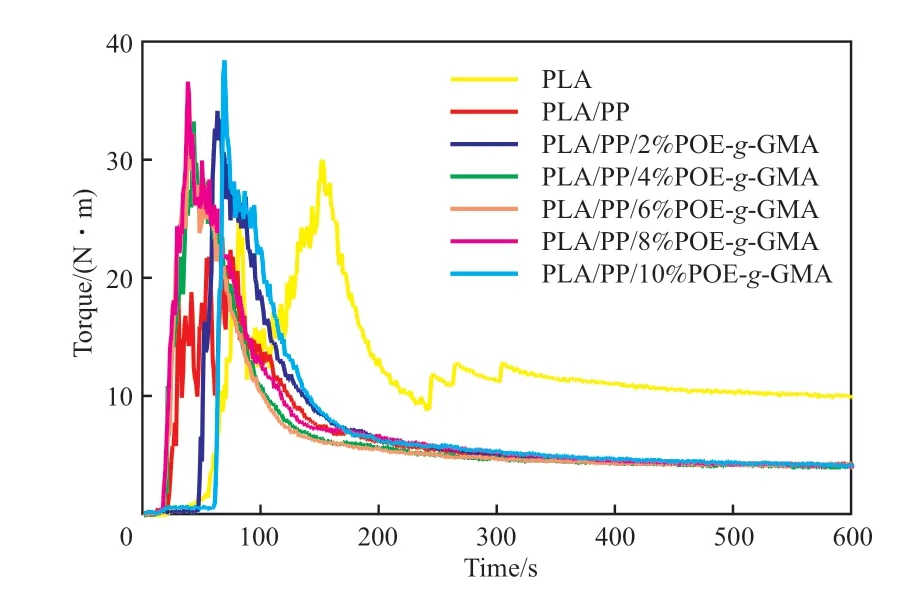
图1 PLA和PLA/POE-g-GMA/PP共混物的扭矩-时间关系曲线Fig.1 The torque-time relationship diagram of PLA and PLA/POE-g-GMA/PP blends.
由图1可知,PLA的平衡扭矩为9.8 N·m,添加20%(w)的PP后,共混物熔体强度降低,平衡扭矩降至4.2 N·m。在此基础上,进一步向PLA/PP共混体系中引入POE-g-GMA,发现随着POE-g-GMA含量的增加,PLA/POE-g-GMA/PP共混体系的平衡扭矩呈先减小后增加的趋势。原因为共混体系中分子量相对较小的POE-g-GMA能够通过增塑作用降低共混体系的熔体强度,但同时POE-g-GMA中的环氧基团能够与PLA的端羟基和羧基通过扩链反应增加熔体强度,在两者的共同作用下,平衡扭矩保持相对稳定。
2.2 POE-g-GMA对PLA/PP共混体系分子间相互作用的影响
图2为试样的FTIR谱图。从图2可看出,PLA在1 180,1 750,2 951,2 999 cm-1处出现特征峰,分别对应C—O—C、C=O、—CH、—CH3的伸缩振动。共混物PLA/PP与PLA的谱图基本相同,表明PLA与PP共混后基团未发生明显变化。在加入相容剂POE-g-GMA后,试样在2 918,2 839 cm-1处出现了POE-g-GMA的特征吸收峰,且各峰面积发生明显变化,表明POE-g-GMA的GMA基团与PLA的端羟基或羧基在共混过程中发生了化学反应。
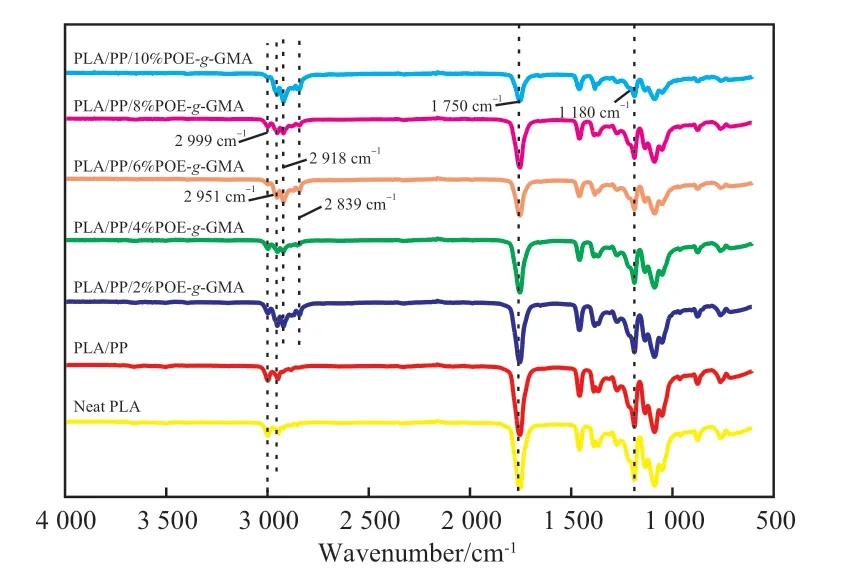
图2 试样的FTIR谱图Fig.2 FTIR spectra of samples.
2.3 POE-g-GMA对PLA/PP共混体系流变性能的影响
POE-g-GMA对PLA/PP共混体系流变性能的影响见图3。从图3a可看出,PLA和PLA/POEg-GMA/PP共混体系的G'随角频率的增大而逐渐增大,主要表现为弹性行为[16]。在低频区,G'随着PP的加入和POE-g-GMA含量的增加而逐渐增加,说明共混物中的分子链的缠结程度增加。这是因为,PP的分子链较PLA链更具弹性,因此更容易缠结,分子链缠结密度提高,导致熔体的可逆弹性变形更高;同时,POE-g-GMA的环氧基团与PLA的端羟基和羧基的反应增加了PLA与PP间的相互作用,从而导致较高的弹性响应。从图3b可看出,POE-g-GMA的引入导致G''逐渐增加,进一步表明了分子链间缠结密度和相互作用增强,导致内耗增加。从图3c可看出,PLA,PP,PLA/PP和PLA/2%POE-g-GMA/PP在低频区的复数黏度变化不大,呈牛顿特性,随后在高频时呈复数黏度降低的非牛顿特性。当POE-g-GMA含量超过2%(w)后,在整个频率范围内,PLA/POE-g-GMA/PP的复数黏度高于PLA,且随角频率的增加而降低,最终趋于平稳[17],即在整个频率范围内均呈非牛顿特性。产生上述现象是因为POE-g-GMA增强了PLA和PP之间相互作用。从图3d可看出,Tanδ(G''/G')表示动态条件下能量损失与能量存储的比值。随POE-g-GMA含量的增加,试样的Tanδ逐渐降低,表明POE-g-GMA改善了PLA与PP的相互作用,共混物的熔融行为得到了增强。

图3 POE-g-GMA对PLA/PP共混体系流变性能的影响Fig.3 Effect of POE-g-GMA on the rheological properties of PLA/PP blends.a Storage modulus;b Loss modulus;c Complex viscosity;d Tanδ
2.4 POE-g-GMA对PLA/PP共混体系结晶性能的影响
图4 为试样的DSC曲线。由图4可知,PLA的玻璃化转变温度(Tg)为60.6 ℃,冷结晶温度(Tcc)为110.6 ℃,结晶度为4%,结晶能力较弱。与PP共混后,PLA/PP相比PLA,Tg变化不大,但Tcc降低,即PLA/PP共混物的结晶能力增强。随POE-g-GMA用量的增大,PLA/POE-g-GMA/PP的Tg与Tcc呈升高趋势,结晶度则先升高后降低。6%(w)的POE-g-GMA即可使共混材料PLA/POEg-GMA/PP的结晶度提升至30.4%,但当POE-g-GMA添加量为10%(w)时,PLA/POE-g-GMA/PP的Tcc升至97.6 ℃,结晶度降至20.7%。原因是低含量的POE-g-GMA具有增塑作用,可提高PLA链段的运动能力,从而提高材料的结晶度;而POE-g-GMA含量过高时,环氧基团与PLA端基反应程度提高,导致在共混体系中形成微交联结构,反而限制了PLA的链段运动,使PLA的结晶能力下降。此外,与PLA/PP相比,PLA/POE-g-GMA/PP的熔融温度升高,说明PLA的晶体更加完善。
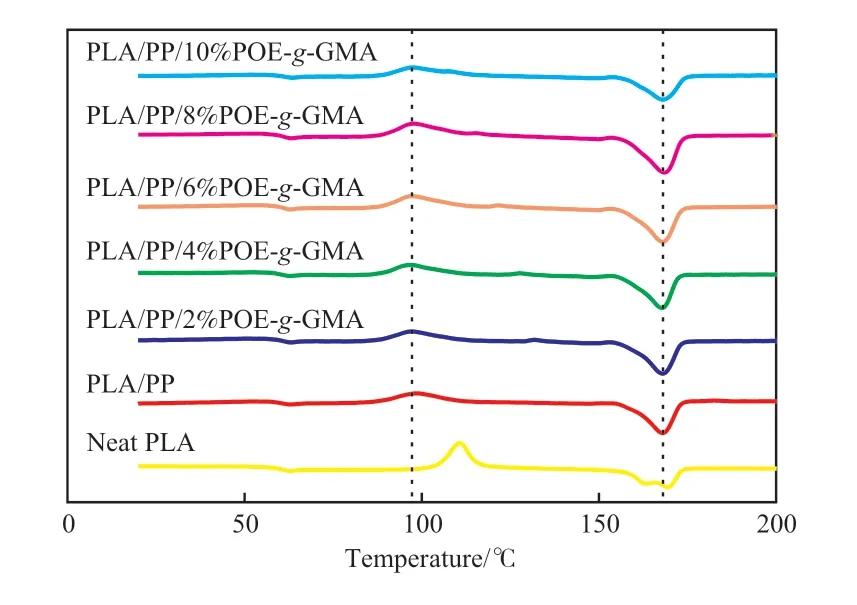
图4 PLA和PLA/POE-g-GMA/PP共混物的DSC二次升温曲线Fig.4 DSC secondary heating curve of PLA and PLA/POE-g-GMA/PP blends.
2.5 POE-g-GMA对PLA/PP共混体系机械性能的影响
图5为试样的力学性能。由图5可知,PLA的拉伸强度为59.9 MPa,但断裂伸长率仅为3.4%,呈明显的脆性。与PP共混后,PLA/PP的拉伸强度与断裂伸长率均下降,说明PP与PLA之间较差的相容性导致PP无法对PLA进行有效增韧。引入POE-g-GMA后,PLA/POE-g-GMA/PP的拉伸强度减小,而断裂伸长率增加。当POE-g-GMA含量为6%(w)时,PLA/POE-g-GMA/PP的断裂伸长率由2.5%提高至25%,较PLA/PP提高了10倍,且拉伸强度达到29.2 MPa,材料的刚韧平衡性良好。当POE-g-GMA的含量超过6%(w)时,共混材料的断裂伸长率反而下降,这是因为过量的POE-g-GMA会在体系中形成团聚,降低链段运动能力,受到外力作用时难以通过链段运动的缓冲而吸收能量,同时POE-g-GMA自身的团聚会产生应力集中,使材料更容易断裂。
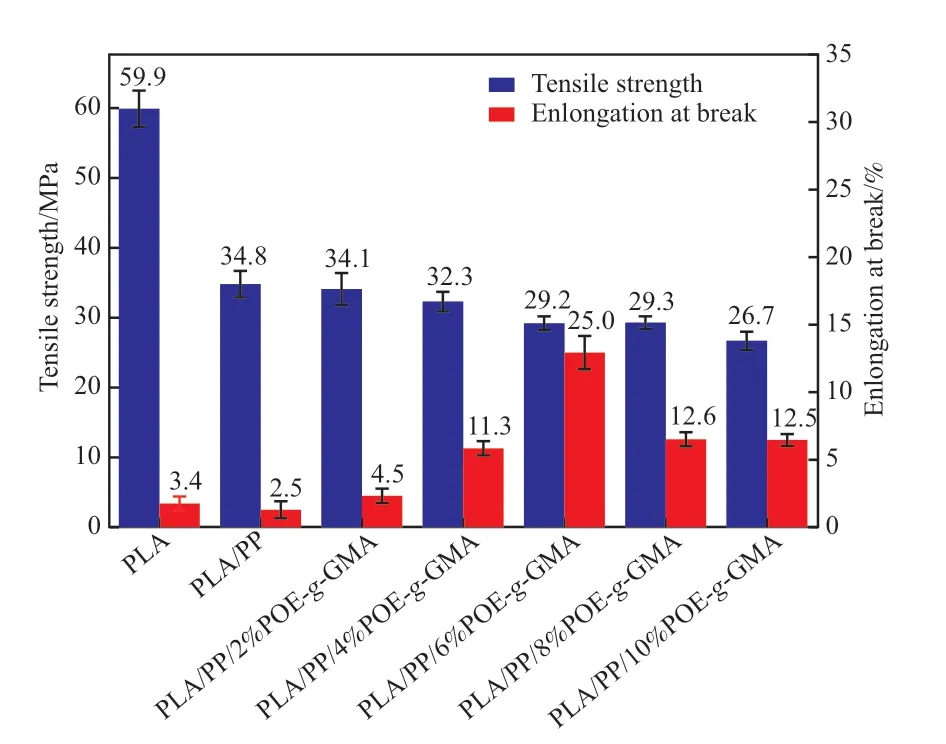
图5 PLA/POE-g-GMA/PP共混体系的力学性能Fig.5 The mechanical properties of PLA/POE-g-GMA/PP blends.
2.6 POE-g-GMA对PLA/PP共混体系界面状态的影响
图6 为试样拉伸断裂面的SEM照片。从图6可看出,PLA断面光滑,为典型的脆性断裂。PLA/PP则呈典型的“海-岛”结构,存在较大尺寸的颗粒及空洞,表明PLA与PP界面相互作用较弱。引入POE-g-GMA后,PP相的尺寸明显减小,且分散均匀,表明PLA与PP相容性有所提高。当POE-g-GMA含量为6%(w)时,PLA与PP的相界面模糊,并发现类似纤维状的结构,表明PLA和PP的相容性较好,界面相互作用较强。POEg-GMA的加入对界面状态的改善机理见图7。从图7可看出,POE-g-GMA中的环氧基团与PLA端基反应,形成微交联结构,使两相结合得更加紧密。继续增加POE-g-GMA的用量,发现共混物相结构由“海-岛”结构向连续相结构转变,但在连续相中发现PP颗粒拔出现象,这是因为过量的POE-g-GMA聚集,导致材料内部发生应力集中降低了共混材料的力学性能。

图6 试样拉伸断裂面的SEM照片Fig.6 SEM image of tensile fracture surface of samples.
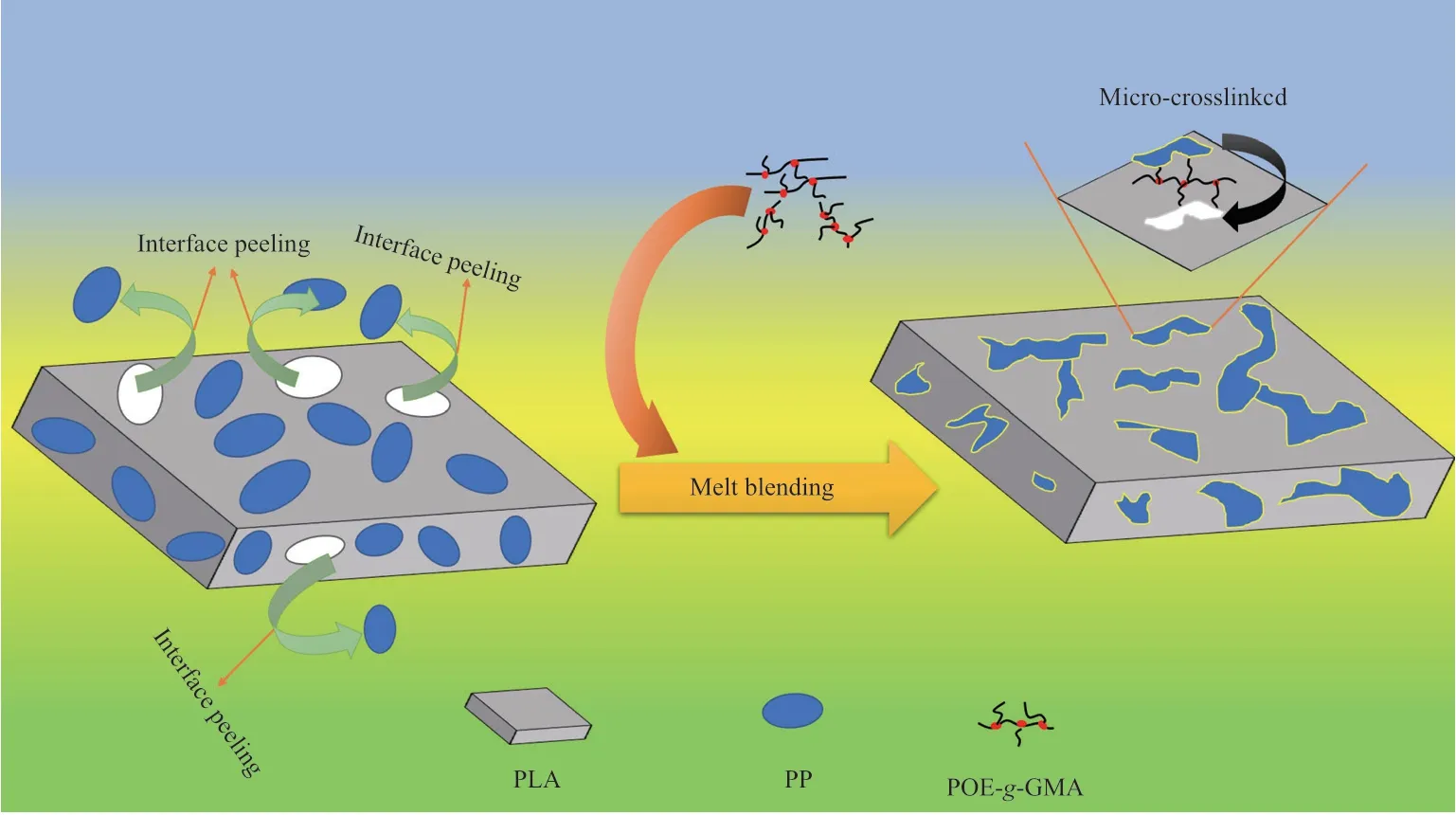
图7 POE-g-GMA对PLA/PP界面调控机理示意图Fig.7 Schematic diagram of the regulation mechanism of POE-g-GMA on the PLA/PP interface.
3 结论
1)POE-g-GMA的GMA基团与PLA的端羟基或羧基在共混过程中发生了化学反应。
2)POE-g-GMA含量超过2%(w)时,PLA/POE-g-GMA/PP在整个频率范围内均呈非牛顿特性。POE-g-GMA改善了PLA与PP的相互作用,使共混物的熔融行为得到了增强。
3)当POE-g-GMA的含量低于6%(w)时,主要起增塑作用,活化PLA分子链的运动能力,提高PLA的结晶度;当POE-g-GMA含量高于6%(w)时,反而降低了PLA的结晶能力,诱导共混体系向连续相结构转变。
4)当POE-g-GMA添加量为6%(w)时,PLA/POE-g-GMA/PP共混材料的拉伸强度达到29.2 MPa,且断裂伸长率提高至25%,为PLA/PP的10倍,材料刚韧平衡性良好。
猜你喜欢
电线电缆(2018年4期)2018-08-31 05:57:30
铜仁学院学报(2018年6期)2018-07-05 09:47:34
电线电缆(2018年2期)2018-05-19 02:03:43
中国科技博览(2017年39期)2017-09-07 09:14:31
核技术(2016年4期)2016-08-22 09:05:24
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
塑料制造(2016年5期)2016-06-15 20:27:39
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01 02:53:54
无机化学学报(2014年8期)2014-02-28 17:32:48
河南科技(2014年8期)2014-02-27 14:07:50
