
Virtual screening of flavonoids from Jatropha gossypiifolia L.as potential drugs for diabetic complications
2022-02-26 03:15YudithCaizaresCarmenateRobertoazAmadorMirthaMayraGonzalezBediaTanTranQuangNhatFranciscoTorrensJuanAlbertoCastilloGarit
Traditional Medicine Research 2022年2期
Yudith Cañizares-Carmenate, Roberto Díaz-Amador, Mirtha Mayra Gonzalez-Bedia, Tan Tran Quang Nhat, Francisco Torrens, Juan Alberto Castillo-Garit
1Unit of Computer-Aided Molecular “Biosilico” Discovery and Bioinformatic Research, Departamento de Farmacia, Facultad de Química-Farmacia, Universidad Central “Marta Abreu” de Las Villas, Santa Clara 54830, Villa Clara, Cuba.2Departamento de Ciencias de la Computación, Facultad de Matemática, Física y Computación, Universidad Celara, Cuntral “Marta Abreu” de Las Villas, Santa Clara 54830, Villa Clara, Cuba.3Institut Universitari de Ciència Molecular,Universitat de València,Edifici d’Instituts de Paterna,P.O.Box 22085,València,Spain.4Unidad de Toxicología Experimental,Universidad de Ciencias Médicas de Villa Clara,Santa Clara 50200,Villa Clara,Cuba.
Abstract Background: Diabetes mellitus is a chronic metabolic disease that is a risk factor for epidemic pathologies.Under hyperglycemic conditions, the enzyme aldose reductase catalyzes the formation of sorbitol in the metabolism of glucose via polyols, leading to the development of diabetic complications.Therefore, inhibitors of this enzyme are therapeutic targets for the prophylaxis and treatment of these conditions.Methods: In this study, a generalized linear regression model was developed to analyze flavonoids- obtained from a database - that have been tested as inhibitors of aldose reductase.In this sense, the molecular descriptors implemented in DRAGON and MATLAB software were used to determine the correlation between the chemical structure of the inhibitors and their pharmacological activity.The model was validated according to the Organisation for Economic Co-operation and Development Standards and subsequently used for the virtual screening of the flavonoids identified in Jatropha gossypiifolia L.Results: The proposed model showed a good fit for its statistical parameters (R2 = 0.95).In addition, it showed good predictive power (R2 ext = 0.94) and robustness (Q2 LOO = 0.92).The experimental chemical space wherein the predictions were reliable (domain of application) was also defined.Finally, the model was used to identify 10 flavonoids from Jatropha gossypiifolia L.as candidates for natural drugs.Compounds with a low probability of oral absorption were identified, among which the elagic acid biflavonoid showed the greatest promise (pIC50 predicted = 9.75).Conclusion: The Jatropha gossypiifolia L.species harbors flavonoids with high potential as inhibitors of the aldose reductase enzyme, in which the biflavonoid ellagic acid was shown to be the most promising inhibitor of the aldose reductase enzyme, suggesting its possible use in the treatment of the late complications of diabetes mellitus.
Keywords: Jatropha gossypiifolia L.; aldose reductase; generalized linear regression model;diabetic complications
Background
The search for therapeutic alternatives, especially those of natural origin, for diabetes mellitus and its complications is the focus of research of many scientists around the world.This chronic metabolic disease prevails worldwide, and has an alarming incidence rate.It is associated with a series of deleterious complications, such as kidney disease, atherosclerosis, and cardiac dysfunction.It affects the main organs, such as the heart, nerves, eyes, kidneys, and blood vessels.In addition, an important element in the current context is its high mortality and morbidity rates combined with the high risk for bacterial or viral infections or cancer development, which make it a major concern with respect to epidemic diseases [1].
Although this disease has been observed from ancient times, with accounts in Egyptian manuscripts dating back to 1500 B.C.E.[2], the first remedies were based on a variety of beliefs and practices of the time, with little understanding of its pathophysiology [3].These natural remedies included diverse and interesting recipes such as rose oil, dates, raw quinces and porridge, snake meat jelly, red coral, sweet almonds, and fresh blind nettle flowers.After studying the sugar content of the urine of the afflicted individuals, the first complete descriptions of this disease were provided by Aretaeus the Cappadocian in the 1stcentury C.E.; he coined the word “diabetes mellitus”, and it was then that “diet and exercise” gained therapeutic value for the control of the disease.Some therapies, such as opium(syrup of poppies), were generously prescribed for the disease for more than 200 years, but they did not treat the disease itself and only relieved certain symptoms caused by complications such as gangrene[4].One of the most significant advances in the treatment of diabetes was the discovery of insulin by the Canadian surgeon Banting and his assistant Best in 1921[5].However,it was not until the 1950s that the first antidiabetic drugs were added to the oral therapeutic arsenal(sulfonylureas).Others compounds, such as metformin, glucosidase inhibitors, and insulin sensitizers, followed during the subsequent decades with different sites of action to allow better metabolic handling and assimilation of the ingested carbohydrates [3].The use of aldose reductase (AR) enzyme inhibitors for the treatment of complications associated with diabetes has recently been studied.This is the limiting enzyme in the polyol pathway that catalyzes the intermediate reaction of sorbitol formation, and its activity increases in hyperglycemic states.The accumulation of sorbitol in tissues is closely related to the occurrence of complications associated with this pathology.Microvascular complications, including retinopathy,nephropathy, and neuropathy, are thought to be especially problematic[6].
Natural products have proven to be an abundant source for the discovery of antidiabetic drugs, with few adverse effects and a low cost [7].Promising secondary metabolites include flavonoids, which act through different mechanisms.One of them is blocking AR enzyme.The exploration of the flavonoid inhibitors of this enzyme constitutes the center of much present research on diabetes therapy, which is directed not only toward the search for new drugs but also toward developing functional foods for diabetics[8, 9].
Traditional spices,herbs,and indigenous plants used throughout the centuries have provided supportive alternatives and potential for future research [3].In particular,Jatropha gossypiifoliaL.is a perennial shrub plant belonging to theEuphorbiaceaefamily and is widely cultivated worldwide as a medicinal and ornamental plant[10-13].This appears in theDictionary of the Various Common Names of Many Common or Notable Plants of the Old and New World, by Dr.Don Miguel Colmeiro, professor and director of the Madrid Botanical Garden in 1871, as Frailecillo de Cuba, Sibidigua, or Tuatúa [14].The traditional uses of this plant have an ancestral origin wherein the Wayuu ethnic group (indigenous to the Guajira Peninsula, on the Caribbean Sea, who live mainly in the territories of the department of La Guajira in Colombia and the state of Zulia in Venezuela) used the latex that comes out of the stem to combat eye conditions [15](therapeutic concoction known as the “eyewash of the Wayuu”) [16].Other traditional uses of this plant include the treatment of burns(thus also known as“burn plant”), leprosy,toothache,eczema, itching,and ulcers,and its use as an antidote for snakebite and as an antibiotic,insecticide, and analgesic, among others.An oil with emetic and purgative, antibiotic, diuretic, febrifuge, abortifacient, and stimulant properties is extracted from its seeds [17].Although this plant is traditionally used against diabetes mellitus in several countries, few studies have scientifically supported this activity and the mechanisms by which it acts [9].In Cuba, the decoction of the whole plant is used in combination withMelia azedarach(aerial parts),Ocimum tenuiflorum(aerial parts),Petroselinum crispum(aerial parts),Solanum americanum(aerial parts),Tecoma stans(aerial parts), andEuropean Tilia(flowers) [18]; in Guinea [19] and Nicaragua [20] the decoction of the leaves is used in combination therapies.However, it was not until the end of the 20thcentury that aqueous decoctions ofJatropha gossypiifoliaL.were reported as antidiabetics in Colombia and the Dominican Republic [21].Later studies showed that ethanolic extracts applied in single or multiple doses to a diabetic rat model showed a reduction in glucose levels [17, 22]; however, the compounds responsible for the pharmacological action were not identified [23].These results show that the methanolic extract of this species is a promising candidate for the development of drugs for the treatment of diabetes and its associated complications [24].Among the compounds identified in this species with potential use as antidiabetics and in the treatment of other conditions, flavonoids stand out.In 2015, Granados et al.determined that a new flavanone, isolated from the leaves ofJatropha gossypiifoliaL., significantly stimulated glucose uptake in C2Cl2myotubes under the conditions of palmitate-induced insulin resistance[23].
The medicinal value of the species has been very controversial, as it is reported to be a toxic plant containing chemicals capable of affecting the nervous, cardiovascular, and digestive systems [25, 26].For this reason, phytochemical studies must focus on the chemical composition of the different plant extracts in order to identify the important compounds involved in its pharmacological actions[13].
QSAR modeling is a useful tool for identifying compounds that possess appropriate physicochemical characteristics for a given biological activity.This analytical methodology starts with the principle that the chemical structure of a chemical compound determines its activity.In other words, it is a process that allows for the establishment of a mathematical correlation between the structure of a compound and its activity [27].In addition, it allows for the prediction of new cases that lack an experimental response, within a short period of time and without a large expenditure of resources on reagents and equipment.In this context, in the present study, we aimed to use QSAR modeling to predict the activity of the flavonoids ofJatropha gossypiifoliaL.species against the AR enzyme to ameliorate a group of diabetic complications that affect the quality of life of the patients with diabetes.
Methods
Database
In this study, we used a database obtained from the study of Boukaraiet al.[28].The AR enzyme inhibitory activities (half-maximal inhibitory concentration is expressed as IC50.) of a set of twenty-nine derivatives of flavones (phenyl-benzopyrane) were analyzed.These derivatives were previously studied and selected because they have been synthesized and evaluated for their in vitro inhibitory activities against AR (in terms of -logIC50, expressed as pIC50).Their chemical structures and in vitro inhibitory activities against AR are shown in Figure 1 and Table 1.
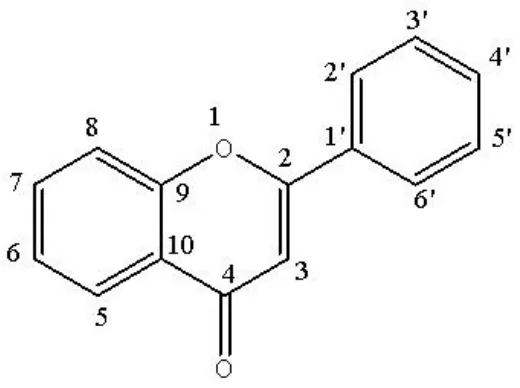
Figure 1 Chemical structure of phenyl-benzopyrane derivatives against aldose reductase
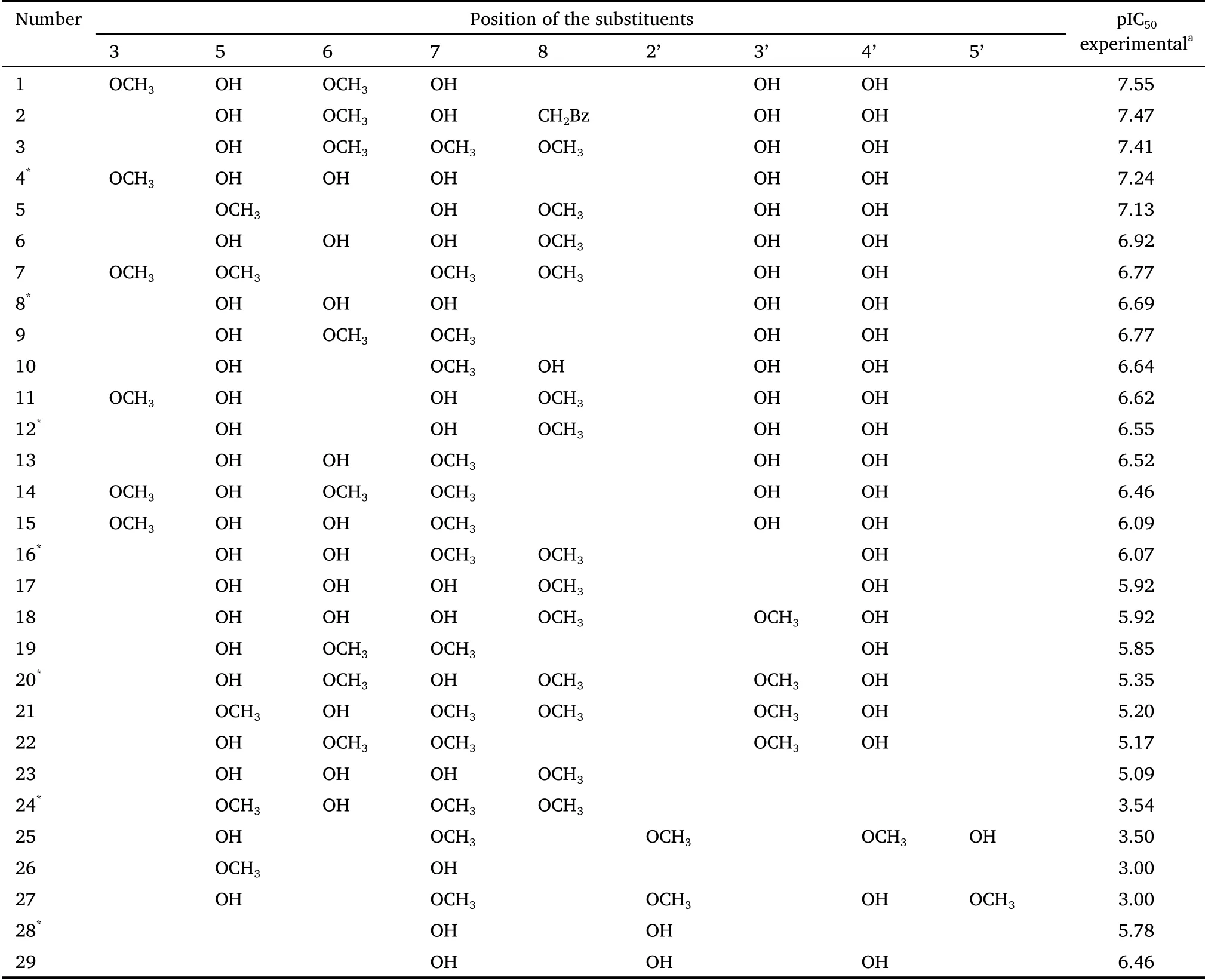
Table 1 Experimental activity of phenyl-benzopyrane derivatives against aldose reductase (IC50 in mol/L)
Of these compounds, 75% were used to train the QSAR model at a rate of 3×1, so that 25% of the flavonoids were left out to make up the external prediction series.It is important to bear in mind that all these compounds are structurally related; that is, they have a common basic nucleus, and their inhibition of AR depends on the substituents at the 3, 5,6, 7, 8, 2’, 3’, 4’,and 5’positions.
Molecular descriptors
In this study, the DRAGON software (version 7.0.10,https://chm.kode-solutions.net) was used to calculate molecular descriptors and transform chemical information into statistically processed numbers [29].This program includes families of constitutional descriptors, descriptors that count substructures and specific functional groups, two-dimensional or topological descriptors and three-dimensional or geometric descriptors.The calculated descriptors were generated starting from a three-dimensional representation of the molecules, previously optimized by the semi-empirical method “Austin model 1”, implemented in Mopac.This process allows for the achievement of a three-dimensional conformation of minimum energy, which is of vital importance for the calculation of three-dimensional descriptors and should not be discarded in enzymatic studies given the stereospecificity of the catalytic site of the enzymes.In addition, in this process, we discarded constant molecular descriptors and those that presented a correlation greater than 95%, as they do not characterize a particular molecule.That is, they are not significant for modeling the inhibition of AR using this database.
Variable selection
The selection of the variables used in this model was undertaken using the MATLAB software (version Matlab2015a, http://mathworks.com)numerical computation system with the sequential method.This method has two components: an objective function and a search method.In this case, the mean square error was used as the objective function, such that the number and combination of variables that minimize the mean square error must be determined.The search method defines whether features are added to each subset to evaluate the objective function, and the forward method was used.Using this combination of the objective function and search criteria, the variables that constitute the predictor model were determined, both qualitatively and quantitatively.It cannot be said that the obtained model was perfect, but it did provide preliminary information regarding the activity of the molecules.
Chemometric analysis
Chemometrics is a chemical discipline that uses mathematical and statistical methods to design or select optimal measurement procedures and experiments and to provide maximum chemical information by analyzing chemical data [30].In this case, an adjusted generalized linear regression technique (generalized linear model,GLM) was applied, taking into account the characteristics of the dependent variable (continuous variable), which was implemented in MATLAB.
The GLM constitutes a special class of nonlinear models that describe the nonlinear relationship between a response (AR inhibition)and predictors (molecular descriptors).A GLM has generalized characteristics of a linear regression model in which the response variable can follow a normal, binomial, Poisson, gamma, or inverse Gaussian distribution with parameters that include the mean response,μ.The link function, f, defines the relationship between μ and the linear combination of predictors.
Model validation
According to the chemometric approach, a model is useful for the detection and selection of chemicals without experimental data if it is carefully verified and a thorough validation is performed [31].Accordingly, we used the Organisation for Economic Co-operation and Development (OECD) principles to validate the proposed model to increase confidence in the reliability of the predictions [32].
Principle 1 is associated with the definition of a measurement point,which refers to a biological property that can be measured and therefore modeled.The objective of this principle is to guarantee transparency at the measurement point predicted by a given model[33].In this study, we used the cytotoxic concentration required to inhibit the AR enzyme by 50% (IC50), expressed as pIC50, as a measure of the activity of the compounds in the database.
Principle 2 establishes that QSAR models must be expressed in the form of unambiguous algorithms, considering that the model algorithm is a way to relate the descriptors of the structure and the chemical activity (measurement point of the model) through mathematical models or rules based on knowledge developed by one or more experts [33].In this case, an adjusted generalized linear regression algorithm was used, as defined in the previous section.
In order to define the applicability domain (AD) of the model(principle 3), the graph of Williams [34] was used, such that the reliable predictions of the model had leverage values lower than the critical leverage, with ± 2.5 standard deviation, and the compounds with values lying outside of these ranges could be considered outliers.
To evaluate principle 4, different exercises were performed.The fit of the model was evaluated according to the coefficient of determination (R2), which must have values close to 1, such that the predicted values correspond to the observed values.
The robustness and stability of the model were verified according to the leaving one out (LOO) criterion in the cross-validation or internal validation.This method allows for a compound to be iteratively excluded from the dataset, and the method then computes a model with the remaining compounds and makes the prediction for the excluded case.If the internal predictions are good, Q2LOO(explained variance in prediction LOO) has a high value comparable to R2and the model is considered to be internally stable or robust.
The ability of the model to predict new cases was verified through external validation.This process was carried out by applying the equation of the model obtained with the training series to a set of prediction data that has never been used in the calculation of the model.If the performance of the model is good, it should have an external prediction set of R2(R2ext)comparable to the R2of the model.
The mechanistic interpretation of the model (principle 5) was carried out based on the selected descriptors to describe the inhibition of AR by a group of flavonoids derived from the flavone phenyl-benzopyrane.
Virtual screening
In this study, only the flavonoids ofJatropha gossypiifoliaL.were considered, although they have been extensively studied and dissimilar metabolites have been characterized, which constitute potential therapeutic candidates.It is undeniable that all metabolites have implicit importance, but the database used was very congeneric and included only flavonoids, so other groups of compounds would be outside the AD of the model, and the predictions for these would not be reliable.The screened flavonoids [23, 35, 36] and the corresponding SMILE codes are shown in the Supporting Material Table S1.
To identify the most promising flavonoids, we proposed that (1)they should be predicted with a high pIC50, (2) they should be within the AD of the model, and (3) they should have drug-like properties suitable for oral administration.To evaluate the third aspect, it was verified that the molecules comply with Lipinski’s rule of five[37], for which the Lipinski alert index (LAI) molecular descriptor was used.
Results
Obtaining the generalized linear regression model
In this study, a GLM is proposed, in which the response variable follows a normal Gaussian distribution.The algorithm of the QSAR-GLM model is as formula (1).
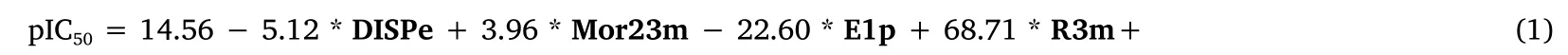
The symbols corresponds to selected molecular descriptors: “DISPe”refers to d COMMA2 value/weighted by atomic Sanderson electronegativities, “Mor23m” refers to 3D-MoRSE signal 23/weighted by atomic masses, “E1p” refers to 1stcomponent accessibility directional WHIM index/weighted by atomic polarizabilities, and“R3m+” refers to R maximal autocorrelation of lag 3/weighted by atomic masses.The parameters are set as follows: N = 22, R2= 0.95,R2adj= 0.90,e= 0.1, where N is the number of compounds in the training set, R2is the coefficient of determination, R2adjis the adjusted coefficient of determination, andeis the mean square error over the estimation in the training set.Figure 2 shows a scatter plot of the predicted versus experimental response.This figure shows the values predicted by the model equation against the experimental pIC50values for the training (blue circles) and prediction (red circles) series.The experimental pIC50values of the phenyl-benzopyrane derivatives for AR and the results predicted by the GLM model in the training set are listed in the Supporting Material Table S2.
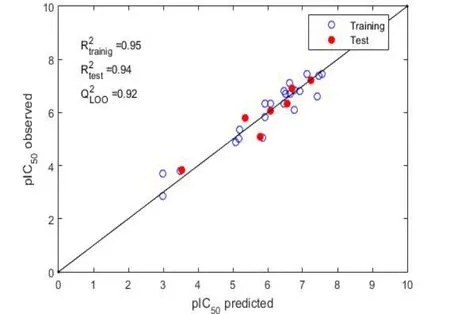
Figure 2 Correlation graph.
Validation
To evaluate the practical utility of the model in predicting AR inhibition, we tested five regulatory principles approved by the OECD for QSAR modeling.Principles 1 and 2 are discussed in the section“model validation”.To verify principle 3, that is the determination of the AD of the model, the leverage and standardized residuals approach described in literature [34] were used.In Figure 3, the Williams plot of the model is shown for the training (blue circles) and prediction(red circles) series.
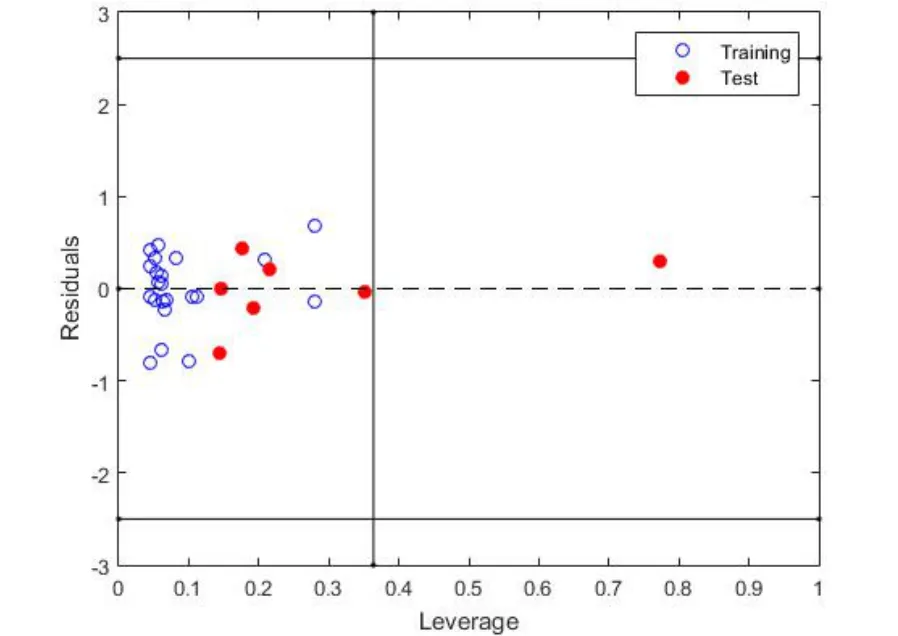
Figure 3 Structural applicability domain of the model (Williams graph).
The robustness of the model (principle 4) was verified by developing internal validation exercises, leaving one case out and obtaining values of Q2LOO= 0.92.The predictive power of the model was tested using an external prediction set with R2ext= 0.94.The predictions made by the GLM model in the prediction set are listed in the Supporting Material Table S3.
Virtual screening of the flavonoids reported in Jatropha gossypiifolia L.
According to the proposed selection criteria, 41 flavonoids present inJatropha gossypiifoliaL.species were evaluated in silico (Table S1).The pIC50values predicted by the model for each flavonoid are listed in Table 2.The model identified 15 flavonoids with pIC50> 6,although only 10 met the requirements for oral administration (Figure 4),and ellagic acid showed the highest activity in silico(pIC50=9.75).
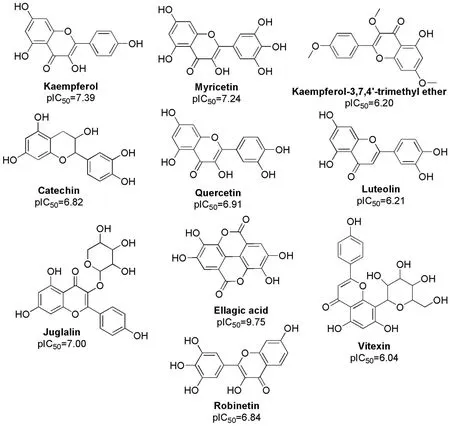
Figure 4 Chemical structure of the most promising flavonoids of Jatropha gossypiifolia L.as aldose reductase inhibitors
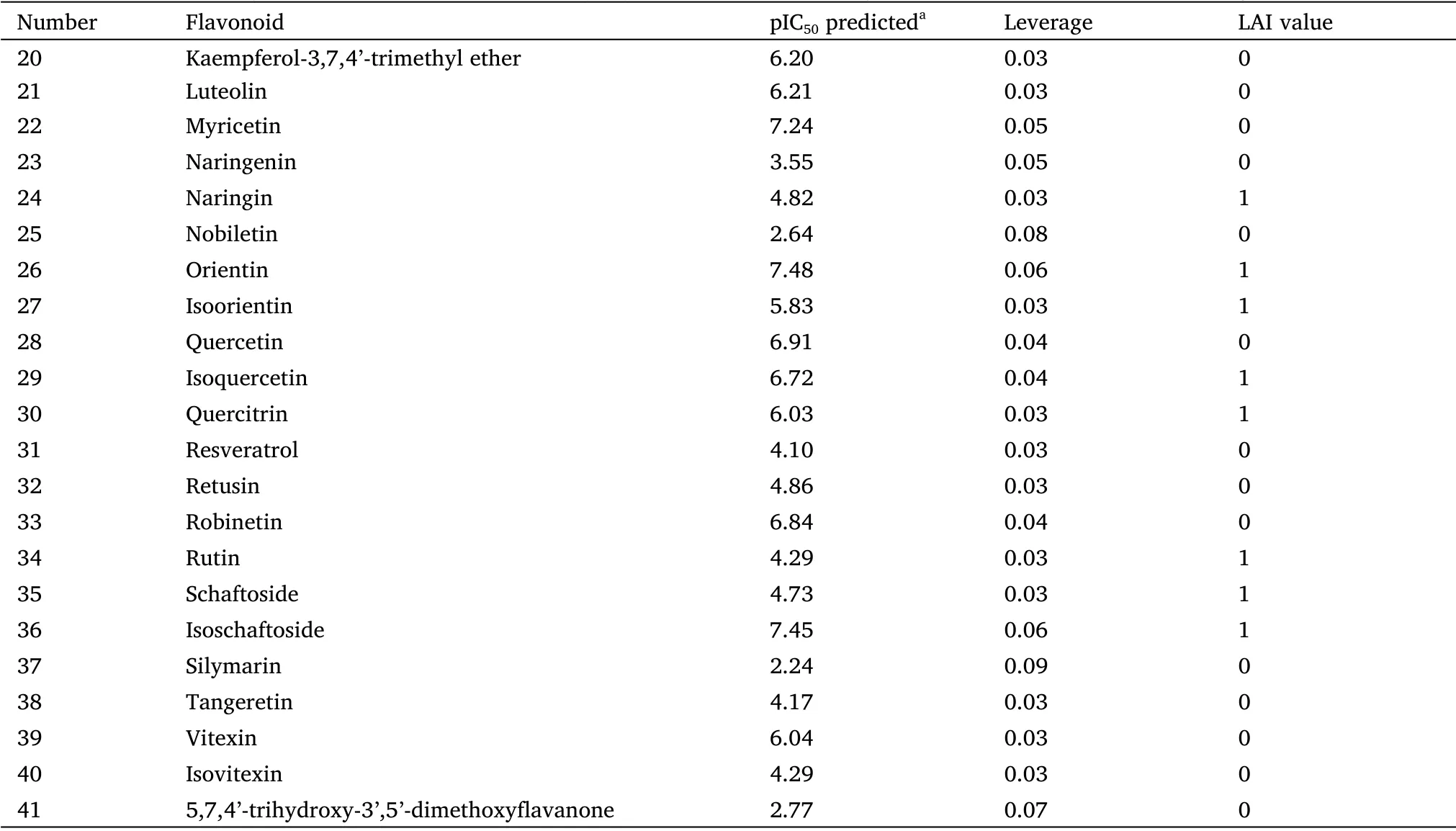
Table 2 Activity of the flavonoids of Jatropha gossypiifolia L.predicted by the model in the virtual screening(continued)
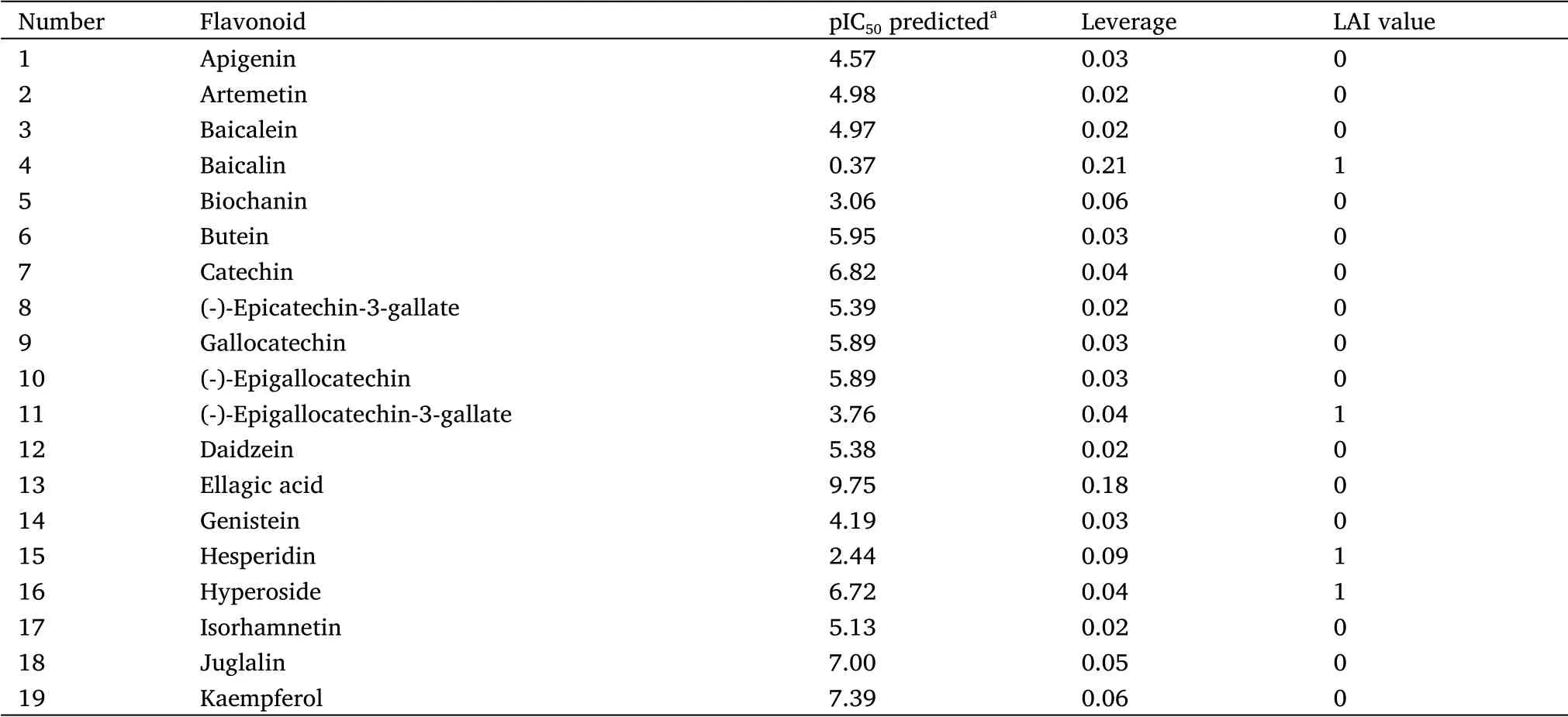
Table 2 Activity of the flavonoids of Jatropha gossypiifolia L.predicted by the model in the virtual screening
The chemical structures of the most promising flavonoids are shown in Figure 4.And 12 compounds did not show an adequate pharmacokinetic profile for the oral route of administration(Supporting Material Figure S1).Furthermore, we define the AD of these flavonoids according to the leverage approach proposed by Gramatica [38].The Insubria graph (Figure 5) shows the compounds in the chemical space of the training series.
Discussion
As evident from the results, the model showed a good fit to model the inhibition of AR by flavonoids derived from the flavone phenyl-benzopyrane (R2= 0.95).It is a simple and easy-to-use model,with only four predictive variables and a small error of the estimate (e= 0.14).The scatter plot of the predicted versus the experimental response (Figure 2) shows that all the observations are near the main diagonal line with low residual values(Supporting Materials Tables S2 and S3).This indicates that there is a good correlation between the selected molecular descriptors and AR inhibition.Furthermore, none of the flavonoids analyzed in the study showed atypical behavior.
The results of the validation exercises showed that most of the compounds were within the AD of the model (Figure 3).There was only one compound in the prediction series with a leverage value higher than the critical leverage, although it showed residuals between ± 2.5 standard deviation - flavonoid_24 (leverage value =0.77 and residual = -0.30).This compound must be considered carefully, as its predictions are not reliable.According to the results obtained during the internal validation, it can be said that the internal predictions were good.A value of Q2LOO=0.92 was obtained,which is high and comparable with R2= 0.95, so the model is considered internally stable or robust.On the other hand, external validation confirmed that the obtained model has good predictive capacity.Its parameters give results adjusted to the R2value of the model (R2ext=0.94), which indicates that it can be used to predict new compounds before their experimental evaluation.The predictions of the external set are shown in the Supporting Material Table S3 (red circles in Figure 2).
Principle 5 is not mandatory because descriptors are usually difficult to interpret because of their mathematical complexity.In this case, the four molecular descriptors of the model are three-dimensional and demonstrate the importance of the specific conformational characteristics of the inhibitors to access the active site of the enzyme and establish interactions with a higher affinity than that of the substrate.In addition, it must be taken into account that the atomic electronegativity (evaluated with the DISPe descriptor), atomic mass (evaluated with the Mor23m and R3m+descriptors), and polarizability (evaluated with the E1p descriptor)largely determine the three-dimensional conformation of the inhibitors and their interaction with the enzyme.According to this analysis, the selected molecular descriptors are suitable for modeling the inhibition of AR by flavonoid compounds.
Once developed and validated, the QSAR-GLM model was used to predict the inhibitory activity of theJatropha gossypiifoliaL.flavonoids against AR.All flavonoids screened were within the chemical space of the training series, as shown in Figure 5.Therefore, they present as drug candidates for the treatment of diabetes complications because of their potential inhibition of the AR enzyme.If these compounds, or even the plant extract rich in the identified flavonoids, inhibit the enzyme, then they could reduce the formation of sorbitol and the presentation of diabetic complications, such as nephropathies,cataracts, atherosclerosis, and cardiac dysfunction, among others.
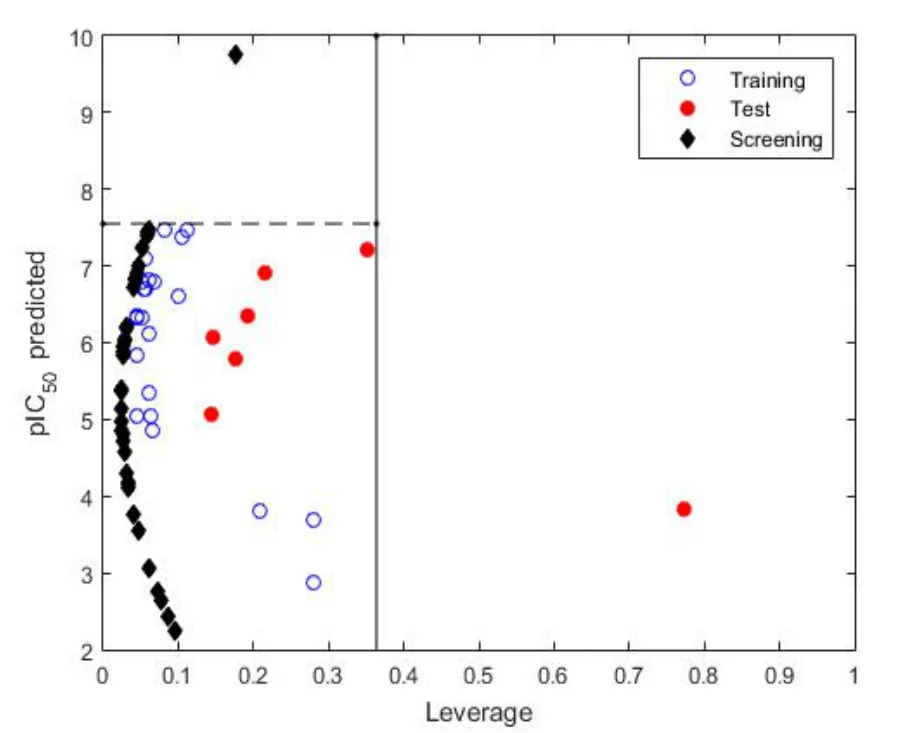
Figure 5 Insubria graph for chemicals without data.
Ellagic acid showed a predicted pIC50value higher than those of all compounds in the database (pIC50= 9.75).It is a polyphenolic acid with proven antioxidant and antitumor properties [39] and has been shown to be the most promising inhibitor of the AR enzyme.This behavior may be due to its evident structural differences (Figure 4).It is a biflavonoid with a more compact chemical structure compared to that of the other screened flavonoids, which suggests that it may be more accessible to the catalytic site of the enzyme.It also contains phenolic groups that allow for the formation of hydrogen bonds with the amino acids of this site, which then favors the inhibition of free glucose binding at the active site, thus, avoiding the production of sorbitol under hyperglycemic conditions [40].Previous studies on the inhibition of AR by this compound, which is present in many dietary sources,indicate that it is a potent inhibitor of AR[40].The high pIC50value obtained in the present study corresponds to that reported in the literature.This demonstrates the high quality and high predictive power of the model proposed in the present study to predict the activity of flavonoid derivatives and can be used to evaluate any flavonoid of natural or synthetic origin, as long as it is within the AD.
On the other hand, it is logical to think that if a treatment is proposed to avoid the complications of a chronic pathology, it should be administered orally.An easy method for evaluating the pharmacokinetic characteristics of orally administered compounds is Lipinski’s rule of five[37].The LAI descriptor takes values of 0 as long as the five criteria are met and it is assumed that the molecules are absorbed in the gastrointestinal tract; it assumes a value of 1 when at least one of the five criteria is violated.In this study, 12 compounds(Supporting Material Figure S1) did not show an adequate pharmacokinetic profile for the oral route of administration.It can be observed that they are flavonoids with a high number of atoms or undergoing a greater degree of glycosylation.Furthermore, flavonoids can be deglycosylated to aglycones by β-glucosidase in the small intestine and are subjected toO-methylation during transfer to the small intestine.However, it must be considered that aglycones can act as pro-drugs and play a role in the pharmacological activity of flavonoids.For this reason, aglycones should not be completely ruled out as therapeutic agents.Further, an exhaustive study on their therapeutic potential should be carried out, if their administration by the oral route is desired or that via the parenteral route is suggested for orally incapacitated patients.
The most promisingJatropha gossypiifoliaL.flavonoids determined in this study are shown in Figure 4.In the case of juglalin (predicted value of pIC50(pIC50predicted) = 7.00), kaempferol-3-O-arabinoside can be modified before absorption to kaempferol (pIC50predicted =7.39) to increase the activity of this flavonoid.However, vitexin (pIC50predicted = 6.04) was modified to its aglycone apigenin (pIC50predicted = 4.57), which decreased its activity, and was therefore not considered to be a good pro-drug.
Using the results of this study as the basis,future continued research can focus on undertaking an in vitro evaluation of the compounds identified with our computational model.In addition, the use of this tool helps avoid performing“trial and error”procedures,consequently,saving much time and money.
Conclusion
The obtained simple and robust QSAR-GLM model could satisfactorily correlate the chemical structure of natural flavonoids with their inhibitory activity against the AR enzyme.The validity of the model according to OECD standards supports its applicability in predicting the activity of new natural or synthetic flavonoids.TheJatropha gossypiifoliaL.species harbors flavonoids with high potential as inhibitors of the AR enzyme, which suggests its possible use in the treatment of the late complications of diabetes mellitus and supports its ethnomedicinal use in diabetes therapy.The biflavonoid ellagic acid was shown to be the most promising inhibitor of the AR enzyme;accordingly, the use of drugs, nutritional supplements, and foods that contain this compound can contribute to the management of the complications of diabetes mellitus.
 Traditional Medicine Research2022年2期
Traditional Medicine Research2022年2期
- Traditional Medicine Research的其它文章
- A comprehensive review of research progress in Chinese medicines for primary liver cancer treatment
- Preparation and characterization of resistant starch type 3 from yam and its effect on the gut microbiota
- Study on technical efficiency of traditional Chinese medicine industry of the Belt and Road Initiative based on environmental complexity
- Yangxin Dawayimixike honey paste inhibits atherosclerosis in ApoE-/- mice by attenuating blood lipids and exerting anti-inflammatory activity
- Traditional herbal medicine as adjunctive therapy for colorectal cancer: a scoping review
- Acupuncture:a new method to treat tic disorders in children