
Light chain cardiac amyloidosis in a nonagenarian
2022-02-26 06:45KojiTakahashiMinaYamashitaTomokiSakaueDaijiroEnomotoShigekiUemuraTakafumiOkuraShuntaroIkedaTakanoriSenbaAkiraSaijoNobuhisaYamamuraSoheiKitazawa
Journal of Geriatric Cardiology 2022年1期
Koji Takahashi✉, Mina Yamashita, Tomoki Sakaue Daijiro Enomoto, Shigeki Uemura,Takafumi Okura, Shuntaro Ikeda Takanori Senba, Akira Saijo, Nobuhisa Yamamura,Sohei Kitazawa
1.Department of Community Emergency Medicine, Ehime University Graduate School of Medicine, Ehime, Japan;2.Department of Cardiology, Yawatahama City General Hospital, Ehime, Japan; 3.Department of Hematology, Yawatahama City General Hospital, Ehime, Japan; 4.Department of Clinical Pathology, Yawatahama City General Hospital, Ehime,Japan; 5.Department of Molecular Pathology, Ehime University Graduate School of Medicine, Ehime, Japan✉ Correspondence to: michitokitatsumasa@gmail.com https://doi.org/10.11909/j.issn.1671-5411.2022.01.008
Cardiac amyloidosis is an infiltrative and restrictive cardiomyopathy caused by the extracellular deposition of amyloid fibrils within the heart as systemic amyloidosis, leading to heart failure, reduced quality of life, and death.[1]There are two major amyloid fibril proteins that affect the heart: amyloid immunoglobulin light chain(AL) and amyloid transthyretin (ATTR).The latter is further subdivided into wild-type ATTR and variant types based on the presence of a mutation in the transthyretin gene.
Systemic AL amyloidosis is a rare multiorgan disease with an incidence ranging from 9.7-14.0 cases/million person-years.[2]In systemic AL amyloidosis, monoclonal light chains are produced by a small B-cell clone.These are deposited in any organ of the body in the form of insoluble amyloid fibrils and lead to organ toxicity.Heart involvement is a major determinant of survival.[1,3]Most systemic AL amyloidosis affects the heart, although approximately half causes heart failure.[4]AL amyloidosis generally occurs in patients aged 60-70 years, and such cases in nonagenarian are very rare.[5-7]
A 92-year-old Japanese male patient was admitted to Yawatahama City General Hospital with a 1-day history of exertional dyspnea.He had no history of smoking or alcohol consumption.His medical history included anemia due to bleeding episodes from internal hemorrhoids 2 years before admission, and urinary incontinence due to benign prostatic hypertrophy since the last year.
Two years prior, the patient was referred to Yawatahama City General Hospital by a general practitioner because of peripheral edema and an elevated serum brain natriuretic peptide (BNP) level[217.8 pg/mL (reference range, ≤ 18.4 pg/mL)].Chest radiography revealed mild pulmonary congestion and the retention of a small pleural effusion.Electrocardiography (ECG) was almost normal;however, the height of the R-waves was reduced in all leads as compared to that 10 years prior (Figures 1A and 1B).Echocardiography demonstrated a normal left ventricular (LV) ejection fraction (EF) with a slight increase in interventricular septum thickness(IVST) (Figure 2A and Table 1).Two-dimensional speckle-tracking echocardiography (2D-STE)demonstrated a near-normal global LV longitudinal strain (GLS) with a normal relative apical longitudinal strain [average apical longitudinal strain/(average basal longitudinal strain + average midlongitudinal strain)],[8]and normal left atrial (LA)reservoir and contraction strains.A small pericardial effusion was also detected.The patient was diagnosed with heart failure with preserved LVEF, of which the trigger was anemia (hemoglobin level,7.9 g/dL).The patient returned to the general practitioner and received treatment for heart failure.
One year prior, the patient was admitted to the Department of Internal Medicine of Yawatahama City General Hospital with left upper limb palsy.Brain magnetic resonance imaging demonstrated multiple acute-phase infarctions (Figures 3A-3C),and he was diagnosed with brain embolism.His serum BNP level was 805.9 pg/mL, and chest radiography revealed mild pulmonary congestion with pleural effusion.An ECG showed low voltage and left axis deviation in the frontal plane (Figure 1C).The height of the R-waves in leads V2-V4 was decreased, but the depth of the S-waves in leads V3-V6 was increased, when compared to those 10 and 2 years prior (Figures 1A-1C).Echocardiography demonstrated a near-normal LVEF with an increase in IVST (Figure 2B and Table 1).LV diastolic dysfunction of grade II was discarded.A small pericardial effusion was also detected.The source was suspected to be a cardiogenic thromboembolism complicated by atrial fibrillation (AF), although no AF was detected till 17 days of admission.Anti-coagulation therapy, but not therapy for heart failure,was initiated.The patient was not consulted by cardiologists, and returned to the general practitioner,by whom loop diuretics were prescribed.
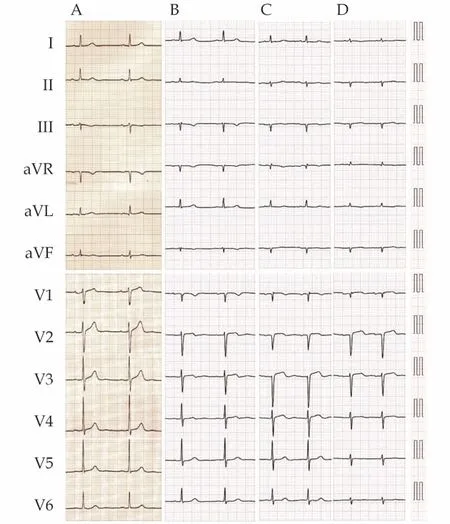
Figure1 Electrocardiogram recorded 10 years before (A), 2 years before (B), 1 year before (C), and on admission (D).The QRS axis in the frontal plane shifted to the left, and the height of the R-waves in both limb and chest leads was decreased.

Table1 Changes over time of echocardiographic findings.

Figure2 Echocardiogram recorded 2 years before (A), 1 year before (B), and on admission (C and D).The interventricular septum thickness (arrows) and the volume of pericardial effusion (asterisks) gradually increased.The right ventricular free walls were also thickened on admission (arrowheads).
On admission, physical examination revealed a body temperature of 36.4 ℃, a systemic blood pressure of 102/68 mmHg, a pulse rate of 81 beats/min,and oxygen saturation of 96% under 1 L/min oxygen administration through a nasal cannula.No cardiac murmurs were audible upon auscultation,although wet rales in the lung fields could be auscultated.The liver was not palpable, and mild pretibial edema was observed.Blood investigations revealed elevated high-sensitivity cardiac troponin I level of 442 pg/mL (reference range, ≤ 34.0 pg/mL)and BNP level of 732.8 pg/mL.Blood count, serum calcium level, and liver function test results were normal.The estimated glomerular filtration rate was 80 mL/min per 1.73 m2and the spot urine protein concentration was 30 mg/dL.Chest radiography revealed mild pulmonary congestion and pleural effusion.ECG revealed left axis deviation,low voltage, and a reduced QRS voltage as compared to those in prior recordings (Figure 1A-1D).Echocardiography demonstrated a reduced LVEF and a thickened IVST (Figure 2C and Table 1).LV diastolic dysfunction of grade III was discarded.Thickened right ventricular free wall and moderate pericardial effusion were also detected (Figures 2C and 2D).2DSTE demonstrated a reduced GLS with a normal relative apical longitudinal strain, and abnormal LA reservoir and contraction strains.The patient was diagnosed with acute decompensated heart failure;thus, tolvaptan was added to loop-diuretics which were prescribed by another hospital.
On day 2 of hospitalization, the patient had rectal bleeding which was confirmed by colonoscopy without achieving a biopsy specimen.The hemoglobin level dropped by 2 g/dL; therefore, anticoagulation therapy was withheld.In addition, the patient had an elemental diet for seven days and did not experience any rectal bleeding.However,anticoagulation therapy remained discontinued thereafter.
The patient was suspected to have cardiac amyloidosis.Technetium-99m-pyrophosphate singlephoton emission computed tomography with fusion to computed tomography images demonstrated no abnormal tracer accumulation in the myocardium (Figure 4).Serum and urine electrophoresis indicated monoclonal immunoglobulin G of type λ.The difference between involved (λ-type)and non-involved (κ-type) immunoglobulin free light chain (FLC) levels (FLC-diff) of 292.2 mg/L was markedly elevated, and the κ/λ ratio of 0.11 was skewed.Multi-parametric flow cytometry analysis of bone marrow plasma cells using CD38 gating strategy demonstrated immunophenotype CD38+CD56+CD19-and an abnormal cytoplasmic immunoglobulin κ/λ light chain ratio of 0.18, implying monoclonality.Monoclonal bone marrow plasma cells accounted for 8.6% of all bone marrow cells.Fluorescence in situ hybridization analysis using the IgH/BCL1 probe did not detect t(11; 14)(q13; q32).Abdominal subcutaneous fat aspiration biopsy, by using Congo red staining and immunohistochemical staining, provided positive findings for λ-type AL amyloidosis.

Figure4 Technetium-99m-pyrophosphate (99mTc-PYP) planar imaging (A, anteroposterior view) and single-photon emission computed tomographic (SPECT) image with fusion to computed tomographic (CT) image (B, horizontal plane at the level of the heart;C, coronal plane) obtained 2 h after injection of the radiotracer.The planar image showed a visual grade 1 cardiac 99mTc-PYP uptake;however, the SPECT image with fusion to CT image showed a blood pool without any evidence of accumulation of the tracer in the myocardium.White asterisks and red asterisks indicate pleural effusion and pericardial effusion, respectively.
Meanwhile, the patient continued to receive cardiac rehabilitation, and continuous ECG from the bedside cardiac monitors showed sinus rhythm.However, he had transient AF complicated with brain embolism on day 24 of hospitalization (Figures 3D-3F).Thereafter, he was found to be gradually emaciated; thus, disease-specific therapies were not administered due to frailty (frailty score 7 assessed by the Clinical Frailty Scale).Finally, he developed aspiration pneumonia with respiratory failure, leading to death.Autopsy of the chest organs revealed a heart weight of 420 g, and amyloid deposits in the bi-atrial/ventricular walls (Figure 5), valves, and conduction system of the heart and pericardium,concomitant with the lung and the esophagus,without any significant atherosclerotic stenoses in the epicardial coronary arteries.In addition, immunochemical classification of the amyloid using antisera directed at the precursor proteins (Figure 5E-5H)and proteome analysis using laser microdissection combined with liquid chromatography-tandem mass spectrometry confirmed λ-type AL amyloidosis and no superimposed ATTR amyloidosis.
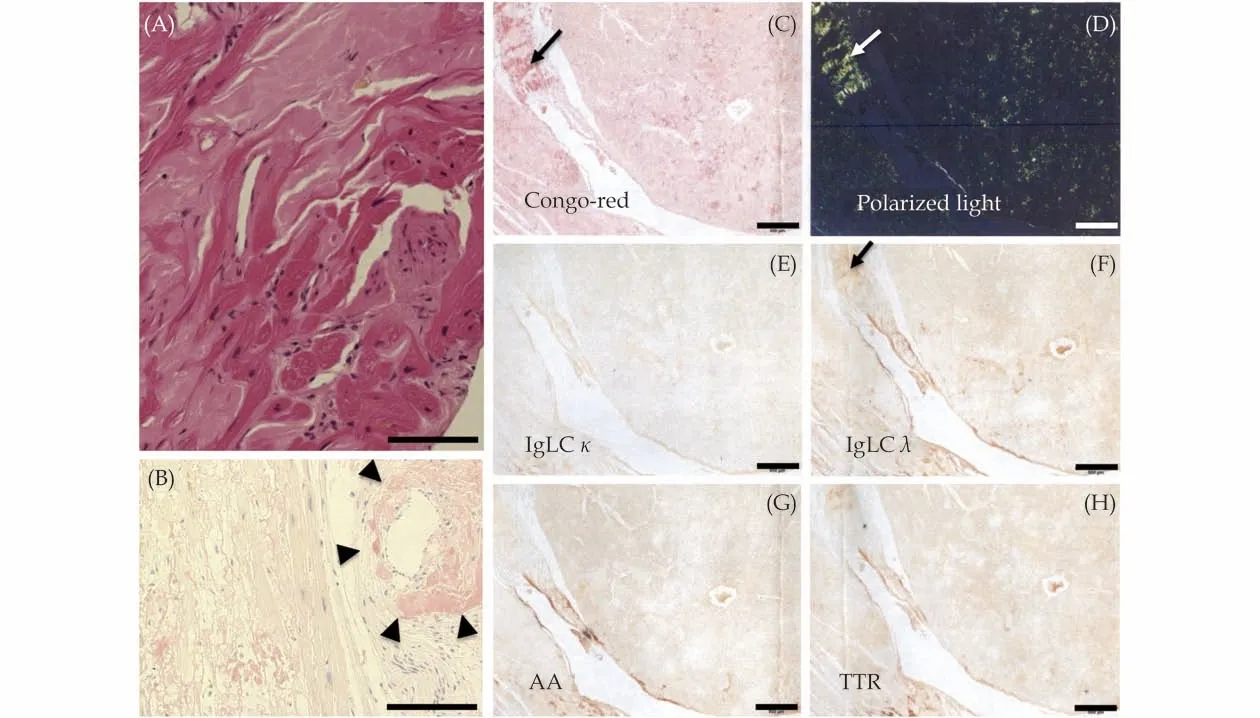
Figure5 Histopathological images of the left ventricular myocardium at autopsy.Panel A (hematoxylin and eosin staining) and panel B (Congo red staining, bright field) showed extensive and extracellular amyloid deposits, compressing the cardiomyocytes (A)and surrounding a small vessel (B, arrowheads).Congo red staining, red-orange under a light microscope (B and C) and apple-green birefringence under a cross-polarized light microscope (D), which are one of the definitions of amyloid deposits, are shown.The panel staining of the amyloid deposits with a set of antibodies raised against each amyloid protein (E-H), i.e., anti-κ light chain (E; IgLC κ),anti-λ light chain (F; IgLC λ), anti-amyloid A (G; AA) and anti-transthyretin (H; TTR) to determine the type of amyloidosis was positive for the antibody raised against the anti-λ light chain and negative for the antibodies raised against other amyloid proteins.Arrows indicate the isolation sites of Congo red-positive amyloid deposits with the laser capture microdissection system before applying them to liquid chromatography-tandem mass spectrometry.The scale bars in panels A-B and C-H indicate 200 and 500 μm, respectively.
AL amyloidosis is diagnosed at ≥ 1 year after the clinical onset in almost 40% of the patients, and this delayed diagnosis often translates into an advanced stage of amyloid organ involvement, limiting access to effective therapies.[7]If untreated, the median survival from the onset of heart failure is approximately 6 months.Our patient with AL cardiac amyloidosis at a diagnosis of 92 years relatively followed a typical course, but the time to establish a diagnosis was protracted.Amyloidogenic light chain-induced cardiac damage is progressive but potentially reversible unless the myocardial lesions are advanced.[4]Thus, the strategy for improving survival in AL amyloidosis is based on early diagnosis while the cardiac damage is still reversible.For that purpose, cardiologists, to whom patients are often referred, should be careful not to miss the opportunity to diagnose AL cardiac amyloidosis by paying attention to “red flags” and by a high index of suspicion.

Figure3 Diffusion-weighted head magnetic resonance images taken 1 year before (A-C), and on day 24 of hospitalization (D-F).High signals (arrows) indicating new infarction are shown as white matter in the right frontal lobe (A) and bilateral pre-central gyrus(B and C), bilateral cerebellar hemispheres (D), the right posterior lobe (E), and the right frontal lobe (F).
In order to favor early and pre-symptomatic detection of amyloid-related organ damage, cardiologists as well as hematologists should promote a periodic yearly simple screening for at-risk patients with monoclonal gammopathy of undetermined significance (MGUS) and an abnormal serum FLC κ/λ ratio, based on the N-terminal pro-BNP level and the presence of albuminuria.[3,4,7]This is because, almost all patients with AL amyloidosis evolve from an asymptomatic pre-malignant stage termed MGUS.[9]An increase in FLC-diff, indicating tumor burden and the degree of involvement of various organs, also precedes the development of the disease for many years in almost all AL amyloidosis patients.[10]In our patient, heart failure with preserved LVEF had developed 2 years prior, although the echocardiography including 2D-STE did not demonstrate typical findings suggestive of cardiac amyloidosis, such as an apical sparing pattern.When cardiologists examine patients with heart failure, particularly whose LVEF is preserved, serum and urine immunofixation electrophoresis, serum FLC analysis, and radionuclide scintigraphy using bone-avid tracer should be performed for suspected cardiac amyloidosis.
It is very interesting that LVEF decreased from 55% to 32% only one year in our patient, suggesting amyloidogenic light chain toxicity.The pathophysiology of AL cardiac amyloidosis differs from that of wild-type ATTR cardiac amyloidosis.Although in both diseases amyloid fibrils deposit within the myocardium, which leads to passive myocardial restriction and dysfunction, clinical observations have suggested that the degree of LV systolic dysfunction in AL cardiac amyloidosis is more severe than that in wild-type ATTR cardiac amyloidosis, despite higher LV mass in the latter.[1,3]Circulating soluble light chains may also be directly toxic to tissues, such as the heart.In addition,marked amyloid deposits surrounding the coronary arterioles in the LV myocardium were detected in our patient.This finding is more prominent in AL cardiac amyloidosis than in ATTR cardiac amyloidosis,[1]and may result in myocardial ischemia with LV systolic dysfunction in the setting of no significant atherosclerosis in the epicardial coronary arteries.Falk,et al.[1]reported that low-voltage ECG often precedes heart failure and may be present before an increased LV wall thickness is apparent on an echocardiogram.This specific ECG finding is an early marker of AL cardiac amyloidosis and is unlikely in ATTR cardiac amyloidosis.[11]The decrease in the height of the R-waves in the ECG recorded 2 years prior, as shown in our patient (Figure 1),might have the same meaning as the above-mentioned finding.
It is worth noting that in our patient, thromboembolism occurred, although AF was not documented one year prior.In both AL cardiac amyloidosis and ATTR cardiac amyloidosis, atrial dysfunction resulting from atrial infiltration with amyloid deposits may cause thrombus formation even in the setting of a sinus rhythm.[1]Thromboembolism may thus be an early manifestation of the disease, and unless the clinician is aware of the phenomenon of LA dysfunction, the source of neurological or systemic embolism may not be recognized.In the setting of an embolic stroke of an undetermined source, which accounts for approximately 17% of all ischemic stroke patients,[12]atrial cardiopathy in cardiac amyloidosis should always be considered as potential embolic sources.In contrast, patients with AL amyloidosis often develop bleeding episodes mainly because of the fragility of the vessel walls due to amyloid deposits.[13]In our patient, rectal bleeding occurred, although a histopathological examination was not performed then.Accordingly, there is a dilemma in the treatment of whether anti-thrombotic therapy is continued.
Autologous stem cell transplantation remains the standard of care for young patients and is generally not adapted to elderly patients.Moreover, recent advances in the management of AL amyloidosis using novel agents to eliminate the underlying plasma cell clone have improved the outcome.[3,14]In our patient living with severe frailty, even when diagnosed at an early stage, treatment options were limited to the management of symptoms without anti-plasma celldirected therapy.Patients who are no longer able to tolerate aggressive treatment may choose to enroll in hospice care.In particular, interdisciplinary palliative care teams should be included in the management of elderly patients with AL amyloidosis as with oncology patients.[15]
ACKNOWLEDGEMENTS
We express our sincere thanks to Dr.Taro Yamashita at the Department of Neurology, Graduate School of Medical Sciences, Kumamoto University,Kumamoto, Japan for performing immunohistochemistry and proteome analysis using autopsy specimens.We would like to thank Editage (www.editage.com) for English language editing.
ETHICS
Informed consent was obtained from the patient or his family members for publication of this case reports and associated images.This case report was writing respecting patient confidentiality and privacy.Family members of the patient had the opportunity to read the present case report and had no objections to the final abstract.
CONFLICTS OF INTEREST
None.
 Journal of Geriatric Cardiology2022年1期
Journal of Geriatric Cardiology2022年1期
- Journal of Geriatric Cardiology的其它文章
- Atrial fibrillation in older adults with cancer
- Vectorcardiographic QRS area as a predictor of response to cardiac resynchronization therapy
- Electrocardiographic markers of cardiac resynchronization therapy response: delayed time to intrinsicoid deflection onset in lateral leads
- Novel electrocardiographic dyssynchrony criteria that may improve patient selection for cardiac resynchronization therapy
- Evolving concept of dyssynchrony and its utility
- Alcohol consumption in relation to the incidence of atrial fibrillation in an elderly Chinese population