
甘草次酸脂质体制备及其药剂学性质分析
2022-02-24 02:48:12张素红李阳杰
化工设计通讯 2022年2期
张素红,周 敬,李阳杰,刘 影
(郑州工业应用技术学院药学与化学工程学院,河南郑州 451150)
作为甘草的主要组成成分,甘草次酸是甘草根部与根茎提炼获得,是五环三萜皂苷类的化合物。其主要功效为抗炎、抗病毒,在防癌、抑制肿瘤方面还具有突出的效果,既往被应用于慢性肝炎及肝癌治疗中取得了较好的效果[1]。甘草次酸与水不相容,采用口服的方式吸收效果差,注射剂的方式治疗生物利用度较低。近年来研究发现,脂质体能够对难溶性药物水溶性起到一定的改善作用,通过对包封药物体内分布的改变,提高治疗靶向性[2~3]。本研究制备甘草次酸脂质体,对其药剂学性质进行分析,旨在为该药物的临床应用提供参考。
1 仪器与试剂
LT3600激光粒度分析仪购自北京海鑫瑞科技有限公司;越众磁力搅拌器数显恒温磁力加热搅拌器购自上海越众仪器设备有限公司;液相色谱质谱联用仪LCMS2000由江苏天瑞仪器股份有限公司提供;微量自动进样器由宁波市镇海玻璃仪器厂提供;Dialysis Membranes透析袋MD25(8000-14000D)购自上海源叶生物科技有限公司;电子显微镜购自麦克奥迪实业集团有限公司;日本奥林巴斯CX23LEDRF(S1/S2)电子显微镜由济南欧莱博电子商务有限公司提供。18β-甘草次酸(18-B-Glycyrrhetinic Acid)购自北京索莱宝科技有限公司,其质量分数在98%以上,规格:20mg支;对照品购自上海雅吉生物科技有限公司,注射蛋黄卵磷脂(Egg Yolk Lecithin)由艾伟拓(上海)医药科技有限公司提供;胆固醇由成都远大化工有限公司提供;十八胺购自盼得(上海)国际贸易有限公司;甲醇为色谱纯,由中山市怡雅生物科技有限公司提供。
2 方法与结果
2.1 试验优化设计
研究通过单因素考察分析及预实验,最终确定甘草次酸脂质体制备变量因素为磷脂∶胆固醇质量(A)、磷脂:药物质量(B)、磷脂∶十八胺质量(C),各因素分别设计3个水平,A因素下比例分别为3∶1,2∶1,1∶1,B因素下设计质量比分别为10∶1,6∶1,2∶1,C试验下设计质量比分别为30∶1,20∶1,10∶1,实施正交试验设计时,对处方的筛选主要参考包封率情况,正交试验结果见表1。
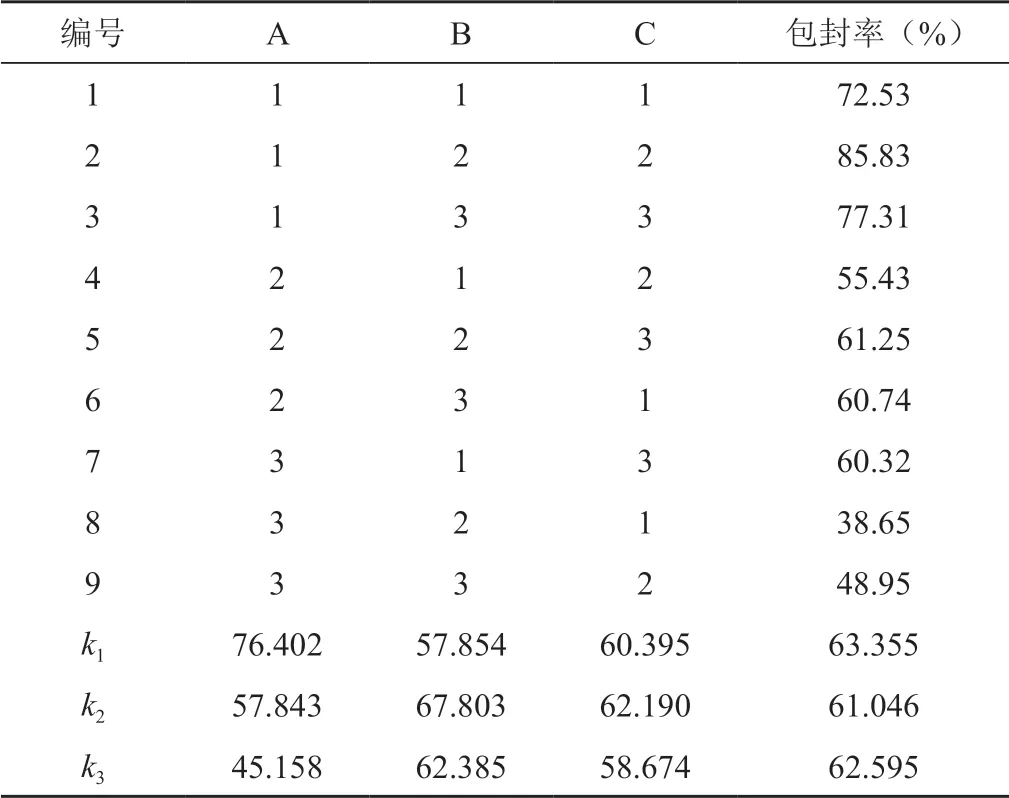
表1 正交试验结果分析
经过方差分析可以发现包封率的影响因素大小为A>B>C,以包封率为考察指标来筛选处方,因此最优组合为A1B2C2,设置A比例3∶1,B比例6∶1,C比例20∶1为最优组合,见表2。
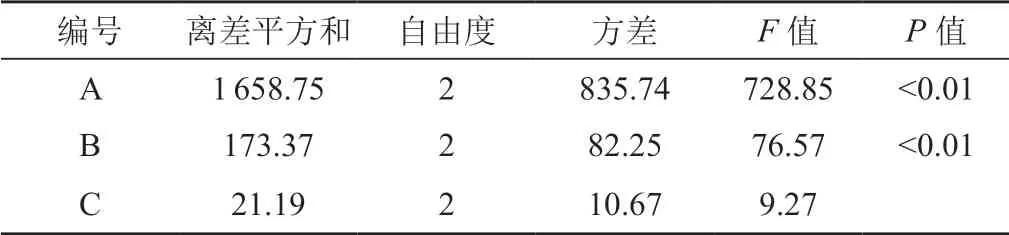
表2 方差分析
2.2 脂质体制备
甘草次酸脂质体制备采用的是乙醇注入法,精密称取磷脂、胆固醇、甘草次酸、十八胺等,将其与无水乙醇混合,获得类脂溶液后,将其加入磷酸盐缓冲液中,需要控制温度为55℃恒温,pH为7.4,在这一过程中需要加入适量的N2将乙醇去除,完成注射后,在45℃环境下孵育,时间以20min为宜,分别在0.8、0.45、0.2µm微孔滤膜获得甘草次酸脂质体[4]。
2.3 脂质体中药测定
2.3.1 色谱条件
选择Thermo赛默飞色谱柱(浙江纳德科学仪器有限公司,型号:Hypersil BDS C18 HPLC 28105-2),规格为250mm×4.6mm,按照89∶10∶1的比例配置甲醇-水-冰醋酸,体积流量以1.0mL/min为宜,设置检测波长为250nm,柱温控制为30℃,以20µL进样量为宜。
2.3.2 专属性试验
将空白脂质体、甘草次酸对照品及脂质体按照处方量配比,加入甲醇溶解,进样后测得的结果如图1所示,可以发现脂质体辅料不会对甘草次酸测定产生干扰。
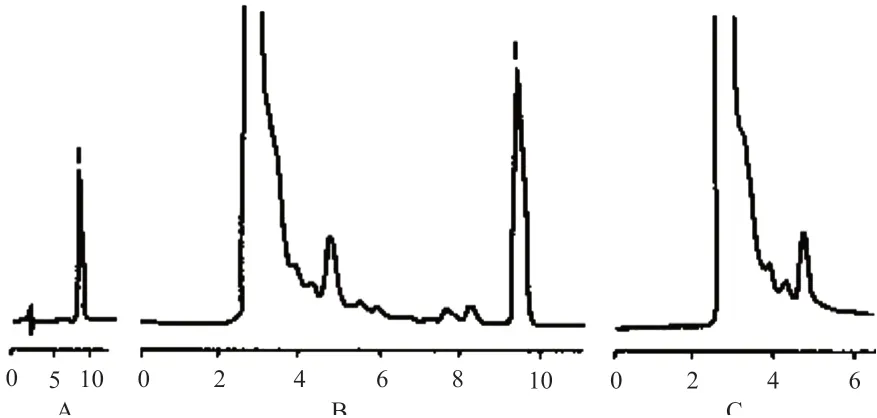
图1 甘草次酸对照品、甘草次酸脂质体、空白脂质体液相色谱图
2.3.3 标准曲线绘制
精密称取甘草次酸对照品,以10.1mg为宜,将其保存于100ml量瓶中,对甲醇进行溶解处理,到达刻度后获得甘草次酸储备液,分别称取0.5、1.0、5.0、10.0、20.0、40.0mL保存在50mL量瓶,定容后获得不同浓度溶液,分别为1.01、2.02、10.10、20.20、40.40、80.80mg/L,进样后对峰面积予以记录,并进行回归分析,回归方程A=0.9284C-0.982,r=0.9997,可以发现在1.01~80.80mg/L范围内线性关系较好。
2.3.4 精密度试验
在对空白脂质体进行制备时,应严格按照处方配比将其加入含有处方药物溶液的量瓶,经过甲醇破乳、稀释定容后,依次分为1.01、10.20、80.80mg/L药物质量浓度,按照2.3.1色谱条件进样并对峰面积、回收率进行计算,计算公式为K1=(A-B)/C×100%,其中A表示加入标准物质的样品测得量,B表示样品中该物质的测得量,C表示加入的标准物质量。按照上述方案及流程制备溶液,测定日内及日间峰面积,对药物质量浓度予以计算,对精密度进行计算,与要求相符[5~6]。
2.3.5 药物测定
在10mL量瓶中加入0.2mL脂质体样品,经过甲醇定容及微孔滤膜过滤,精密称取20µL,对脂质体药物浓度予以计算,可以发现甘草次酸质量浓度平均为(4.77±0.21)ng/L。
2.4 包封率测定
在Sephadex G-50柱上加入0.2mL甘草次酸脂质体,洗脱液作用下对游离药物进行分离。前25mL排除不用,仅收集游离部分,以50mL为宜,经过蒸干处理后,采用流动相对残渣进行溶解、定容,以10mL为宜,经微孔滤膜过滤,提取20µL进样,代入方程式中进行计算。另外在10mL量瓶中置入0.2mL甘草次酸脂质体,加入甲醇后震荡破乳,定容,过滤后,提取20µl进样测定[7]。包封率计算方法为(1-W游离/W总)×100%。经过试验分析,可以发现正交试验下获得包封率平均为(91.53±2.34)%。
2.5 Zeta电位与粒径测定
先稀释甘草次酸脂质体混悬液,脂质体粒径与Zeta电位采用激光粒径分析仪测定,结果发现其粒径平均值为(142±12)nm,带正电,电位为(35.62±5)mV。
2.6 形态观察
稀释甘草次酸脂质体混悬液,采用1%磷钨酸进行复染,经过透射电镜作用对形态予以观察,如图2所示,其外观为大小均一的球形、类球形。
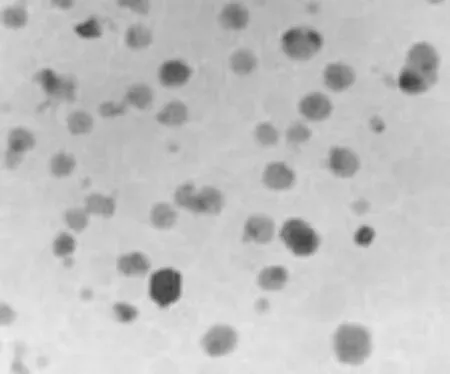
图2 甘草次酸脂质体电镜图片
2.7 体外释放度及稳定性测定
在透析袋中分别放入甘草次酸原料溶液与脂质体混悬液,均为3.0mL,扎紧两端,在搅拌浆上固定,释放度测定pH为9.0,温度以37℃为宜,每分钟100r,取样3mL对同体积空白介质进行补充。经过过滤、进样对峰面积予以测定。可见药物呈缓释放,脂质体在前10h共释放30%,之后保持该速率较为稳定地在72h内缓释,甘草酸溶液释放仅需10h。在考察稳定性方面,先将甘草次酸脂质体混悬液分为3批,保存于4~6℃环境下,时间以2个月为宜[8]。试验开始时、开始后1个月、2个月末取样,观察各批样本在质量分数、外观及粒径等方面的变化情况[9~10],可以发现甘草次酸脂质体从外观上看几乎无变化,质量浓度无改变,试验开始时包封率为90.56%,粒径为143.68nm,1个月末包封率及粒径分别为88.53%、150.25nm,2个月末分别为86.73%、156.72nm。由上述数据可知,低温环境下长时间放置并不会对甘草次酸脂质体外观及质量浓度等产生影响,说明其稳定 性好。
3 结束语
研究基于传统乙醇注入法制备技术,引入正电荷脂质十八胺,采用1:6药脂比例,制备的甘草次酸脂质体具有较高的包封率及较好的稳定性,甘草次酸结晶及脂质体积未发生积聚,且包封率会随着十八胺用量的增加呈现出升高趋势。该工艺合理、稳定,呈缓释特点,能够获得满意的脂质体稳定性。通过该试验能够为甘草次酸脂质体的药物动力学研究提供参考,不断开发新型制剂,为临床主动靶向治疗提供可靠的参考。
猜你喜欢
昆明医科大学学报(2022年3期)2022-04-19 13:59:46
山东化工(2018年15期)2018-09-20 08:55:34
中成药(2018年2期)2018-05-09 07:20:08
中成药(2017年3期)2017-05-17 06:08:52
中成药(2017年3期)2017-05-17 06:08:48
山东医药(2015年16期)2016-01-12 00:40:04
首都食品与医药(2015年18期)2015-11-03 05:59:08
天然产物研究与开发(2014年8期)2014-04-27 14:16:25
亚太传统医药(2012年2期)2012-11-01 16:31:49
药学研究(2012年2期)2012-10-25 05:54:40
