
改性聚苯硫醚纤维高温氧化行为变化机理研究
2022-02-24 03:13:34连丹丹刘伟方尹立新MTRasnatunFerdous卢建军
棉纺织技术 2022年2期
连丹丹 刘伟方 尹立新 MT Rasnatun Ferdous 卢建军
(1.太原理工大学,山西晋中,030600;2.江苏恒力化纤股份有限公司,江苏苏州,215226)
聚苯硫醚(以下简称PPS)纤维具有优异的热稳定性、耐腐蚀、阻燃性能[1-2],近年来在环境保护、化学工业和军工等领域中得到广泛应用。凭借自身优异性能和良好的性价比,PPS纤维已成为国际公认的燃煤电厂高温尾气除尘滤袋的主要使用原料。但是常规PPS纤维在高温下易氧化、交联,强度损失高达30%,致使PPS纤维滤袋综合性能降低,在现有工况下,使用寿命降低[3]。成为制约PPS纤维及其滤袋发展和推广应用的瓶颈。目前,被学者普遍认同的PPS纤维抗氧化改性的方法主要有抗氧剂法和纳米复合材料改性法,但针对PPS纤维氧化前后结构和机理的研究还较少。前期研究中[4],作者通过纳米Ti—SiO2改性PPS纤维(以下简称A-PPS),提高了纤维的耐氧化性能和使用寿命。在前期研究的基础上,根据“国家三部委及中科院等单位联合制定关键材料升级换代工程实施方案”中节能环保产业发展急需新材料的要求,即PM2.5过滤材料长期使用温度不低于240℃的要求,对改性前后PPS纤维的240℃高温氧化做了系统性研究,考察了改性前后PPS纤维的耐氧化能力并对其氧化反应机理进行了分析。
1 试验
1.1 试验原料
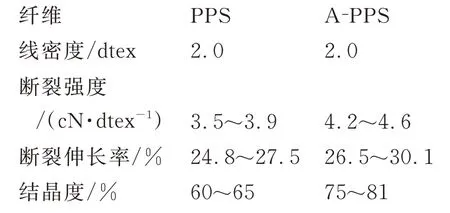
采用溶胶-凝胶法原位改性制备得到的纳米Ti—SiO2,纳米颗粒尺寸在100 nm左右,Ti元素以Ti—O—Si网络结构存在于纳米颗粒结构中,使纳米Ti—SiO2含有大量不饱和残键和活性官能团。试验用纤维均为熔融纺丝法自制所得。A-PPS纤维加入质量分数为1.0%的自制纳米Ti—SiO2。PPS纤维及A-PPS纤维的基本性能如下。
1.2 试验方法
分别取PPS纤维和A-PPS纤维各3份,每份5 g,置于底部铺有玻璃纤维的瓷盘中待用。将烘箱升温至240℃,待温度稳定后将瓷盘置于烘箱中层,保持烘箱与外界通风。高温处理24 h、192 h和360 h后取出待用。
1.3 测试与表征
利用YG004型单纤强力仪测试不同处理时间纤维的强伸性能,每个试样测试50次。
利用美国PekinElmer公司的DSC6000型差式扫描量热仪测试纤维的热学性能。将纤维试样剪碎至粉末状,每个试样精确称量5 mg,置于铝坩埚并压盖。程序以20℃/min由30℃升温至320℃,稳定5 min消除热历史,再以20℃/min降温至30℃,氮气气氛,流量20 mL/min。
利用TD-3700 X型射线衍射仪测试纤维的晶体状态。将粉末状纤维平铺在载物凹区并压平,Cu Kα,波长0.154 nm,扫描步宽0.05°,扫描角度5°~50°,工作电压30 kV,工作电流25 mA。
使用美国PekinElmer公司FT-IR Spectrometer型傅里叶红外光谱仪对纤维样品进行红外光谱表征。使用其ATR部件进行衰减全反射红外光谱扫描。光谱扫描范围4 000 cm-1~650 cm-1,扫描8次。
X射线光电子能谱分析采用AXIS ULTRA DLD(Shimadzu Kratos)能谱仪,X射线源Al Ka(能量1 486.6 eV),测试功率150 W,结合能用C 1s(284.6 eV)标定。
2 结果与讨论
2.1 高温氧化后纤维的外观色泽及强伸性能
PPS纤维及A-PPS纤维高温氧化处理后的外观变化及强伸性能见图1和图2。

图1 PPS纤维及A-PPS纤维高温氧化处理后颜色变化
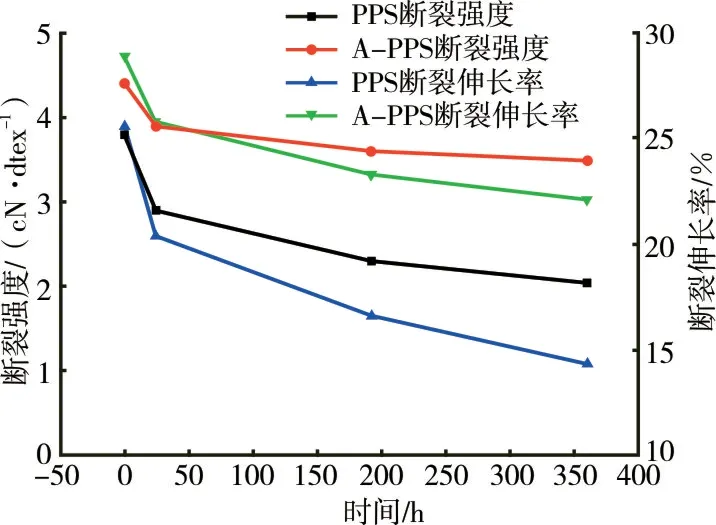
图2 高温处理时间对PPS和A-PPS纤维强伸性能的影响
由图1可见,随着时间的延长,两种纤维的颜色均泛黄,且颜色加深,表明两者均出现氧化的现象。对比相同时间PPS纤维和A-PPS纤维的颜色外观可见,PPS纤维的颜色较A-PPS纤维深,表明相同的条件下PPS纤维的氧化程度高于APPS纤维。
由图2可见,两种纤维的断裂强度和断裂伸长率均随着高温氧化时间的延长而降低,但降低程度差异较大。当时间为360 h时,PPS纤维的强度保持率为53.6%,而A-PPS纤维的强度保持率为79.5%。表明PPS纤维在240℃高温环境中极易氧化造成强度损失。A-PPS纤维的强度保持率较PPS纤维显著提高,高温抗氧化性较优。
2.2 高温氧化后纤维的聚集态结构转变
强度和颜色的改变表明纤维经过高温氧化后大分子结构及聚集态结构发生了转变,首先利用差示扫描量热仪和X射线衍射仪对PPS纤维及A-PPS纤维的聚集态结构进行分析,结果见图3。
由图3可见,随着高温氧化时间延长,改性前后PPS纤维的熔融峰温均向高移动,熔融热焓提高,熔程逐渐缩短。需要注意的是,当高温时间达到360 h时,PPS及A-PPS纤 维 的 熔 点 分 别 为296.3℃和289.8℃,熔融热焓分别为89.98 J/g和83.45 J/g,均已超出PPS的原有范围;且两种纤维的降温结晶初始温度和结晶峰温均随着高温时间的延长而降低。熔融和结晶的变化表明,较长时间的高温氧化不仅能够引起PPS纤维在聚集态结构层面产生变化,更引起了大分子结构的变化。当时间为24 h时,PPS及A-PPS纤维的熔点和熔融热焓分别为281.9℃、278.9℃和71.67 J/g、66.10 J/g,虽然还均在PPS原范围内,但PPS纤维的焓值已经明显偏高,表明24 h的短时高温仅使A-PPS结晶度有提高,但已经使PPS纤维产生了分子结构变化。上述结果表明APPS纤维受高温氧化引起的分子结构变化程度较PPS纤维低,验证了上文颜色和强度的变化。
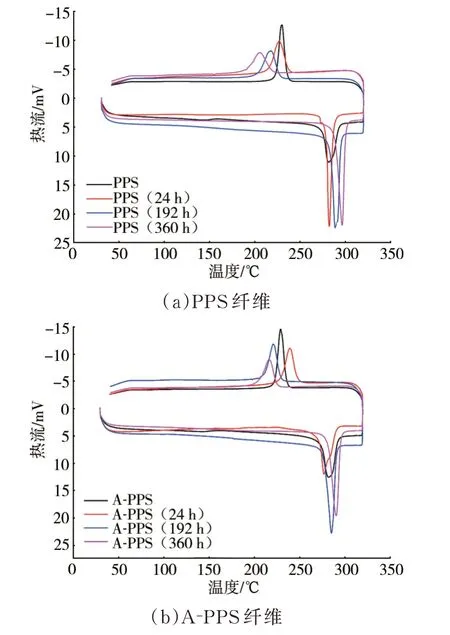
图3 纤维经不同高温氧化时间后的差示扫描量热曲线
不同时间高温氧化后,PPS及A-PPS纤维的X射线衍射图谱见图4。根据文献,PPS晶胞参数为a 0.867 nm,b 0.561 nm,c 1.026 nm,大分子链中的S呈锯齿状排列在平面(100)上,相邻两个苯环与晶面(110)以±45°交替排列,晶格长度与C—S—C键角呈线性关系[5-7]。
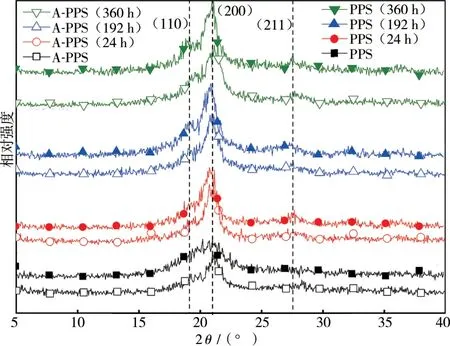
图4 纤维不同高温氧化时间后的X射线衍射图谱
由图4可见,随着时间延长,PPS晶体的(110)面、(211)面和(200)面衍射强度均加强,且峰位高移,如高温时间达360 h时PPS纤维高移0.6°,由2dsin θ=nλ可知,高温氧化后PPS纤维的晶格常数变小,这是由于氧化不但使S氧化成亚砜和砜,并引起了苯环间发生交联,增大了晶格密度。对比图4中A-PPS纤维高温氧化前后X射线衍射峰强和峰位可见,峰值稍有增强且位移微弱,佐证了A-PPS纤维的氧化程度较弱。
2.3 高温氧化后PPS纤维的分子结构变化
由上文分析结果可见,不同时间的高温氧化会引起PPS纤维的聚集态结构发生不同程度的改变,而聚集态结构的转变一方面与大分子链间产生变化有关,另一方面也与大分子链内的变化直接相关。因此,有必要针对大分子链作进一步表征分析,具体见图5。图5中1 060 cm-1~1 040 cm-1亚砜特征峰表明,PPS纤维本身存在一定程度的氧化情况,随着高温氧化时间的延长,硫砜键特征峰显现且逐渐增强,表明纤维大分子链中S元素氧化加强。同时,对比A-PPS纤维红外光谱,高温后同样出现了砜基的特征峰,但强度偏弱。
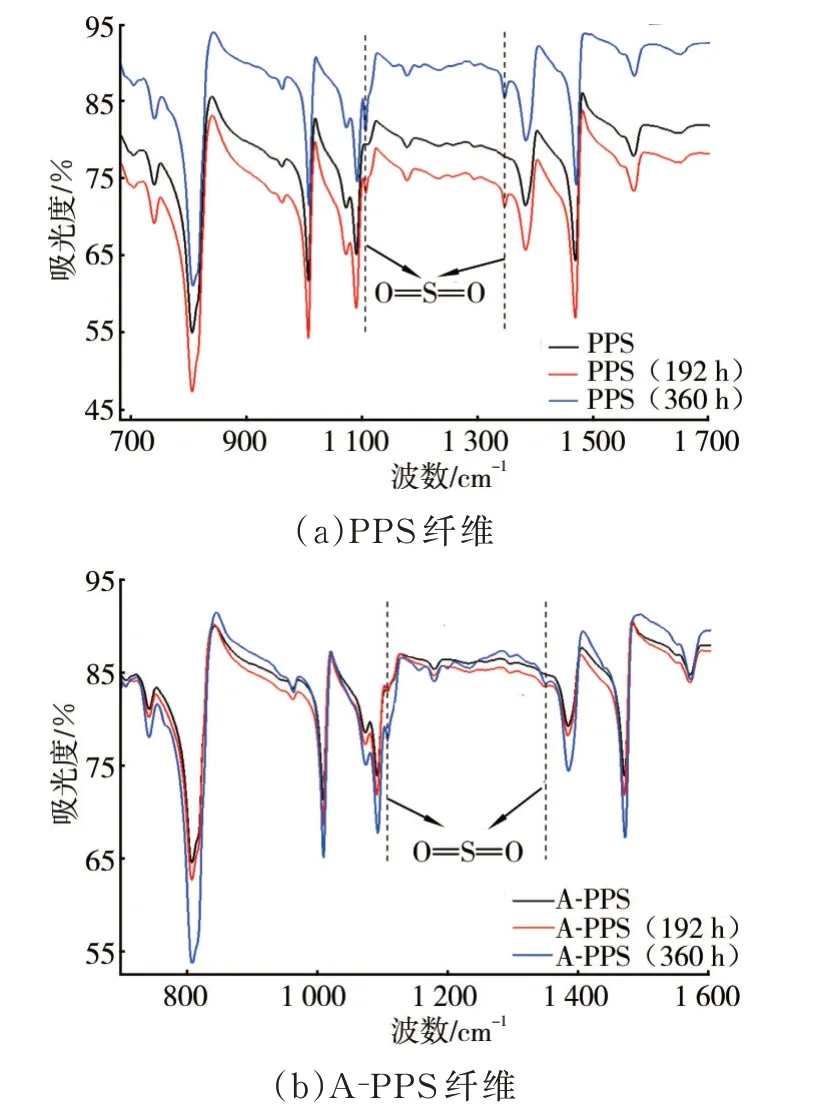
图5 纤维高温氧化后的红外光谱
由于红外光谱本身的扫描范围和精度有限,对650 cm-1以下的特征峰无法进行精确的表征和分析,而大分子苯环间的交联特征峰大部分在625 cm-1~450 cm-1区间,且PPS自身红外特征峰较多,官能团出峰位置存在相互覆盖和干扰,压制了许多高温后应该出现的特征峰,故利用X射线光电子能谱进一步分析PPS纤维高温氧化前后的分子结构变化,结果见图6。由于A-PPS纤维中纳米Ti—SiO2的质量分数仅有1.0%,所以宽谱中Si的特征峰不是很明显。
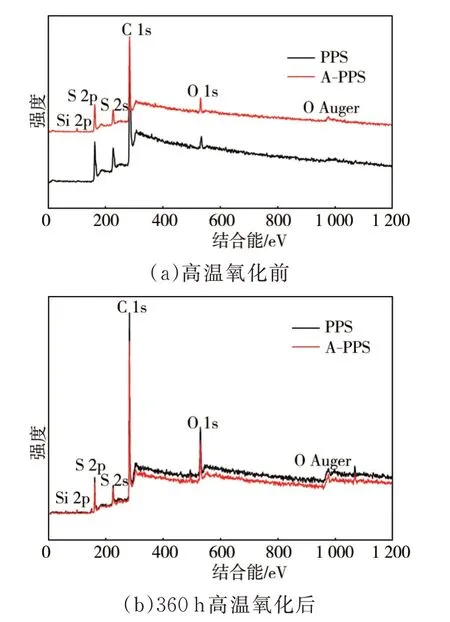
图6 纤维高温氧化前后的X射线光电子能谱
表1为C 1s的高分辨谱和分峰拟合结果。拟合系数在2.0~3.8之间,拟合度较高。
前期的研究结论表明,A-PPS纤维中的纳米Ti—SiO2经过高温高压的纺丝过程后会与PPS大分子链间形成S-Si配位体系和芳香氢键,从而提高A-PPS纤维的抗氧化能力和抗交联能力。由表1可见,高温氧化后芳环C—H键和C—S键含量均减少,芳环交联含量提高,表明高温不但会使PPS纤维大分子链间产生大量的芳环间交联,还会导致C—S键断裂,进而产生交联。对比分析PPS与A-PPS的区别可见,A-PPS纤维本身分子内交联程度较PPS纤维低,高温后交联含量虽然也有一定提高,但主要对应的是芳环C—H和芳香氢键含量的减少,表明交联主要来自大分子链间而非PPS大分子链断裂,这得益于芳香氢键和S—Si配位体系的保护。

表1 纤维高温前后C 1s高分辨谱分峰拟合结果
由于X射线激发下产生自旋-轨道耦合作用,S 2p层电子会在X射线光电子能谱的高分辨谱中以2p3/2和2p1/2双峰形式出现[8-10],表2为S 2p3/2和S 2p1/2劈裂双峰拟合结果。

表2 纤维高温前后S 2p高分辨谱分峰拟合结果
由表2可见,两种纤维高温后在高结合能位均出现较为明显的特征峰,表明氧化产生砜。PPS纤维高温氧化后C—S—C键含量减少,亚砜含量减少,表明分子结构上一方面是大分子链断裂,断裂后硫醚被氧化成砜,另一方面是原有的亚砜被氧化成砜,且从含量的变化上可知这种分子结构上的变化较为明显。对比A-PPS纤维高温前后的变化可见,其氧化程度较PPS纤维要小,这也验证了上文C 1s的分析。
3 结论
(1)改性后PPS纤维的抗氧化性能得到一定程度的提高。
(2)高温氧化会使PPS纤维的结晶热焓和熔融峰温提高,芳环间交联和S的氧化虽没有改变PPS的晶型,但会使PPS本身的晶格常数缩小,晶格密度提高。
(3)氧化后PPS产生大量的芳环间交联,C—S键断裂,分子中S被氧化成亚砜和砜,由于A-PPS纤维中Si—S配位和芳香氢键的存在,能够提高改性后的PPS纤维的抗氧化作用。
猜你喜欢
中学化学(2024年5期)2024-07-08 09:24:57
中学生数理化·八年级物理人教版(2023年10期)2023-11-30 01:57:54
机电安全(2022年5期)2022-12-13 09:22:26
科学(2020年1期)2020-01-06 12:21:34
中学生数理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
中学化学(2016年10期)2017-01-07 08:37:06
浙江大学学报(理学版)(2016年6期)2016-12-15 03:15:03
湖南城市学院学报(自然科学版)(2016年2期)2016-12-01 04:06:40
分析测试学报(2015年3期)2016-01-13 06:18:12
中国药业(2014年17期)2014-05-26 09:07:45
