
组蛋白修饰在成骨细胞中的调控作用
2022-02-19 10:37邝小华赖仲宏刘午阳
赣南医学院学报 2022年12期
邝小华,赖仲宏,刘午阳
(1.寻乌县人民医院骨科;2.赣南医学院第一附属医院骨科,江西 赣州 341000)
骨是一种动态组织,通过骨形成和骨吸收不断进行重塑。负责骨形成的是成骨细胞,它源于骨间充质干细胞(Bone mesenchymal stem cells,BMSCs),其分化和矿化受多种细胞因子和生长因子的调控。越来越多的研究发现组蛋白修饰和成骨细胞分化密切相关。组蛋白存在于真核生物的细胞核中,与DNA组成核小体,是染色质的基本结构。每一个核小体都是由组蛋白H2A、H2B、H3和H4各两分子组成核心八聚体,外面缠绕着长147个碱基对的DNA,在核心八聚体之间由长60个碱基对的DNA连接,该处DNA再连接一个单体组蛋白H1[1]。组蛋白修饰一般发生在H3和H4的尾部,在酶促反应下进行动态修饰,包括甲基化和去甲基化、乙酰化和去乙酰化、磷酸化和去磷酸化等,导致染色质重构和基因启动子的暴露或隐藏,影响基因转录[2]。本文对组蛋白修饰在成骨细胞分化中的作用进行综述,希望为确定相关骨骼疾病的治疗靶点和研发药物提供理论基础。
1 成骨细胞分化的分子调控
BMSCs是一种常见的前体细胞,具有自我更新和分化为成骨细胞、软骨细胞和脂肪细胞的潜能。BMSCs的分化受多种信号的调控(图1),包括经典Wnt信号、转化生长因子-β、骨形态发生蛋白(Bone morphogenetic protein,BMP)以及一些关键转录因子,如RUNX2、osterix、CCAAT增强子 结 合蛋白(CCAAT enhancer binding protein,C/EBP)和过氧化物酶体增殖物激活受体γ,这些信号都是BMSCs分化为前成骨细胞所必需的[3]。MSH Homeobox2(MSX2)、碱性磷酸酶(Alkaline phosphatase,AKP)和Ⅰ型胶原α1(Collagen type I α1,COL1A1)表达增加标志着前成骨细胞形成[4]。Osterix在成骨细胞分化全程都发挥了不可替代的作用。Osterix通过激活骨唾液酸蛋白(Bone sialoprotein,BSP)和骨钙素(Osteocalcin,OC)基因以及上调COL1A1来触发前成骨细胞分化为成骨细胞,并进一步诱导SOST、DMP1和PHEX的表达,为最终形成骨细胞和矿化提供物质基础[5]。
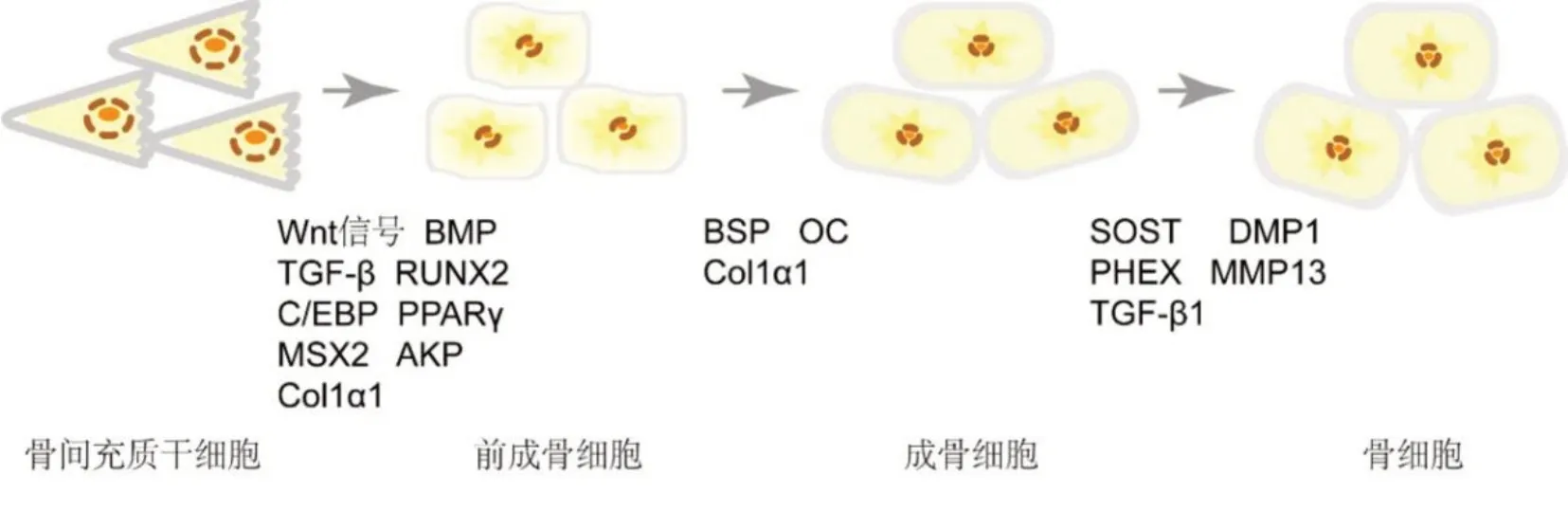
图1 成骨细胞的分化过程
2 组蛋白修饰对成骨细胞的调控
2.1 组蛋白甲基化对成骨细胞的调控 组蛋白甲基化可以发生在赖氨酸和精氨酸残基上,在甲基转移酶的作用下,增加一个、两个或三个甲基的过程。不同位点的氨基酸甲基化有不同的特性,H3K4、H3H36、H3K79的甲基化松解染色质的状态,转录活跃,而H3K9和H3K27的甲基化使染色质更加浓缩,转录沉默[6]。另外,高的甲基化水平功能改变更显著。组蛋白甲基转移酶根据催化结构域序列,可分为SET结构域和非SET结构域。SET结构域是组蛋白甲基转移酶的重要结构域,诱导它们的酶促活性,包括SET家族、SUV家族、EZH2等。
研究表明[7],SETD2可介导其靶基因脂多糖结合蛋白的H3K36me3的富集,维持其染色质的活跃状态,积极进行转录起始和延长,在体内外调节BMSCs向成骨细胞转化。这意味着SETD2及其下游靶基因可能成为代谢性骨病的有效靶点。EZH2基因编码的是一种赖氨酸甲基转移酶,抑制EZH2的活性后发现H3K27me3的高甲基化水平得到了控制,进而刺激成骨细胞成熟,并使用抑制剂GSK126进行了验证[8]。EZH2抑制成骨的关键是BMP2信号的中断,并且EZH2抑制剂和BMP2的联合治疗可刺激BMSCs的成骨分化,同时减少BMP2单独使用的高成本和不良反应问题[9]。因此表观遗传调控和骨合成药物的协同应用可作为改善骨生成的治疗策略。
2.2 组蛋白去甲基化 组蛋白去甲基化是氨基酸残基上的甲基在去甲基酶的催化下脱落的过程。去甲基酶包含赖氨酸特异性脱甲基酶(Lysinespecific demethylase,LSD)和JMJD家族两大类。LSD1是单胺氧化酶,利用FAD依赖机制使H3K4和H3K9去甲基化[10]。LSD1在成骨细胞中高表达,其抑制会损害成骨分化和骨结节形成。在机制上,LSD1与RUNX2的启动子结合,保持H3K4低甲基化水平,是正常成骨分化所必需的[10]。JMJD家族蛋白可选择多种底物,但又有特异性,不同的成员可以催化不同的赖氨酸残基去甲基化。JMJD3可以特异性催化H3K27me3的去甲基化,一方面通过降低抗凋亡蛋白Bcl-2的甲基化水平,激活Bcl-2的转录而减少成骨细胞凋亡,另一方面催化RUNX2和osterix启动子区H3K27me3的去甲基化,增强其转录活性,诱导成骨细胞分化;另外,研究还发现,敲低JMJD3后蛋白激酶D1减少,使促凋亡蛋白Bim的磷酸化程度下降,并增加其活性,促进成骨细胞凋亡[11-12]。KDM4A和KDM7A是新型的成骨细胞分化的去甲基化因子,它们都可与分泌性卷曲相关蛋白4(Secreted frizzled related protein 4,SFRP4)、C/EBPα的启动子结合,并分别靶向去除H3K9me3和H3K9me2、H3K27me2的甲基化,进而上调其表达,另外还能中断经典Wnt信号传导,以促进脂肪生成和抑制成骨[3,13]。这些发现可能有助于为骨质疏松症等代谢性骨病提供新的治疗线索。
2.3 组蛋白乙酰化 组蛋白乙酰化是组蛋白尾部N-端的赖氨酸残基进行乙酰基的加成,这一反应由组蛋白乙酰转移酶催化。乙酰化去除了赖氨酸残基上的正电荷,降低了组蛋白与DNA之间的亲和力,从而打开了浓缩的染色质结构,有利于暴露基因启动子,并且允许转录因子和RNA聚合酶Ⅱ进入DNA,促进基因表达[6]。赖氨酸乙酰转移酶包括P300/CBP、PCAF/GCN5和myst基团。
甲状旁腺激素(Parathyroid hormone,PTH)参与调节骨重塑和钙稳态相关基因的转录。在PTH的刺激下,蛋白激酶A被激活,导致基质金属蛋白酶-13(Matrix metalloproteinase-13,MMP13)的启动子RD/Runt位点的RUNX2蛋白磷酸化,进而大量募集P300,随后MMP13的组蛋白H4和H3相继发生乙酰化,促进基因转录[14-15]。MMP13的表达主要局限在骨骼发育和重塑区域,参与正常骨生长发育和细胞外基质的降解等。另外,TGF-β1也具有和PTH相同的作用机制[16]。因此,刺激PTH和TGF-β1的分泌,改变组蛋白的乙酰化修饰以促进基因表达可能是诱导骨生长的潜在手段。
2.4 组蛋白去乙酰化 组蛋白去乙酰化则是在组蛋白去乙酰化酶(Histone deacetylase,HDAC)的作用下使乙酰基脱落,导致染色质呈现压缩状态,降低启动子活性,沉默基因表达,这一过程也参与调控成骨细胞分化。HDAC家族根据结构、酶活性、在细胞中的位置以及与酵母的序列同源性分为四类:Ⅰ类HDAC(HDAC1,2,3,8)、Ⅱ类HDAC(HDAC4,5,6,7,9,10)、Ⅲ类HDAC(Sirtuin1-7)、Ⅳ类HDAC(HDAC11)[17]。
2.4.1 Ⅰ类HDACs Ⅰ类HDACs定位于细胞核,但不能直接与DNA结合,因而它们通常与一些转录因子和蛋白质组成复合体被招募到染色质而发挥作用,比如SIN3、NuRD、COREST、PRC2和MiDAC等[18]。
在成骨细胞分化过程中,HDAC1表达降低,成骨标志性基因如osterix和骨钙素的启动子处组蛋白H3和H4高度乙酰化,表达显著上调并促进成骨[19]。HDAC3在成骨细胞分化的各个阶段都有高表达,并已被证明对骨量的维持和获得以及正确的颅面骨发育至关重要。HDAC3敲除的小鼠模型显示骨长度缩短、骨量和矿化减少、患有严重的骨质疏松,并遭受频繁的脆性骨折[20]。HDAC3在病理过程中也参与调控,颅咽管瘤组织发生钙化会增加手术切除的难度和风险。HDAC3在钙化的瘤组织中表达增加,但它的核易位受到miR-181b和CBX4的调节。上调的miR-181b直接靶向抑制CBX4的表达,使得CBX4对BMP2介导的HDAC3核易位的抑制作用受 到 限 制,随 后RUNX2、OCN、osterix、OPN和AKP显著增多,促进成骨和钙化[21]。HDAC3也能作为RUNX2的抑制因子调节成骨分化。用HDAC3选择性抑制剂MI192处理BMSCs细胞,检测到HDAC活性降低和组蛋白乙酰化程度增加,并能显著上调成骨细胞相关蛋白,如RUNX2、AKP、COL1A1和OCN,诱导BMSCs向成骨细胞分化和细胞外基质沉积以及矿化,这表明利用表观遗传来增加骨量,治疗成骨不足的相关代谢骨骼疾病具有很大的潜力[22-23]。
2.4.2 Ⅱ类HDACs Ⅱ类HDACs分布于细胞核和细胞质,它们具有一个调控的N-端结构域,确保它们与组织特异性转录因子和共抑制物相互作用。在这类HDACs中,HDAC4与骨形成非常密切,并且和RUNX2之间存在负调控关系。研究表明,TGF-β诱导因子同源盒2(TGF β-induced factor homeobox 2,TGIF2)在BMSCs和原代颅骨成骨细胞中过表达,促进了p-Smad3和HDAC4的相互作用,从而降低了RUNX2乙酰化的丰度,抑制体外成骨细胞分化[24]。
2.4.3 Ⅲ类HDACs Ⅲ类HDACs也称为Sirtuins家族,包含7个成员,它们的结构与其他HDACs大不相同,而且它们的活性依赖NAD+,具有保守的催化结构域和可变的N端和C端结构域[18]。SIRT1是研究最为广泛的,它在骨重建中的重要作用已被报道。研究表明,在胚胎期,SIRT1基因敲除小鼠体型发育较小,存在骨矿化不足和颅骨骨缝闭合缺陷;在老年期补充SIRT1可减少骨骼和肌肉质量的损失,延缓衰老,抑制炎症反应,还促进了成骨细胞分化和矿化[25-27]。机制上,SIRT1有效上调Wnt/β-catenin和BMP2信号通路,这两个通路都是BMSCs和成骨前体细胞向成骨细胞分化的关键,SIRT1上调也能增加骨钙素和RUNX2的表达,促进骨的形成和矿化,并抑制脂肪生成[28]。
3 组蛋白修饰重编程
组蛋白修饰在成骨细胞分化中的作用不容忽视,因此组蛋白修饰重编程可能成为治疗骨代谢疾病具有前景的手段。研究表明[8,29],靶向抑制EZH2能有效降低H3K27me3水平,削弱了它转录沉默的作用,促进BMSCs向成骨细胞分化。纳米钛技术也能参与组蛋白修饰重编程,通过阻碍H3K27me3的积累以暴露成骨细胞标记基因的启动子区域,并保护成骨细胞免受破骨细胞的影响[30]。值得注意的是,脂肪细胞和成骨细胞都是由BMSCs分化而来,因此它们之间的反式分化可能是一种潜在的骨再生方法。脂肪细胞是临床环境中最丰富且最容易获得的细胞,因此,它们可能是反式分化的良好细胞来源。研究证明[31],使用HDAC抑制剂TSA诱导组蛋白H3K9的高度乙酰化,可以增强脂肪细胞分化为成骨细胞的可塑性。
4 小结与展望
组蛋白修饰,尤其甲基化和乙酰化在正常骨形成和骨重建中都扮演了重要的角色,而其他修饰如磷酸化、SUMO化、糖基化等在成骨细胞分化中的作用仍是知识空白。另外,组蛋白乳酸化是近几年提出的全新的翻译后修饰,为成骨细胞的分化提供了新的研究方向。研发组蛋白修饰重编程相关的药物和技术是临床上治疗骨代谢疾病的策略之一,但是面临着提高有效性和选择性的挑战。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
畜牧兽医学报(2022年3期)2022-03-30
中国畜牧兽医(2022年1期)2022-02-15
口腔医学(2021年10期)2021-12-02
现代泌尿外科杂志(2019年10期)2019-10-31
生物学通报(2019年2期)2019-06-15
中华老年口腔医学杂志(2016年2期)2017-01-15
中国组织化学与细胞化学杂志(2016年4期)2016-02-27
中国病理生理杂志(2015年8期)2015-12-21
天津护理(2015年4期)2015-11-10
