
基于语义特征向量的DNA与转录因子结合特异性预测
2022-02-19 10:23孙晓雨权丽君黄立群
计算机应用与软件 2022年2期
孙晓雨 权丽君 梅 杰 黄立群 吕 强*
1(苏州大学计算机科学与技术学院 江苏 苏州 215006) 2(江苏省计算机信息处理技术重点实验室 江苏 苏州 215006)
0 引 言
表达是结构基因在生物体内的转录、翻译及加工的过程,而大量TF与基因组的作用能够影响基因表达[1],其中DNA与特定TF蛋白的结合,即DNA序列特异性,对于转录和选择性剪切这类基因的调控过程研究具有特别重要的意义。由于数据量扩大和特征维度增加,通过生物实验手段获得DNA序列特异性费时费力,因此,目前只有很少一部分的TF和DNA结合特异性得到明确的表征。近年来,随着高通量基因组测序数据的发展和计算性能的提高,类似于ChIP-Seq[2-3]等实验产生的高通量数据对于使用计算手段预测DNA序列特异性、继而寻找发现新的生物学机制起到了至关重要的作用。
目前,多种基于序列的计算方法已经被提出来预测TF与DNA的结合特异性,包括以权重矩阵为代表的传统计算方法和以机器学习为代表的新型学习方法等。位置权重矩阵(Position Weight Matrix,PWM)[4]和二核苷酸权重矩阵(Dinucleotide Weight Matrix,DWM)[5]描述了每一个碱基在每一个位置上出现的频率。它们的不足在于假定每个或每两个核苷酸对TF与DNA序列的结合贡献是独立的,这种粗暴的可加性假设给预测准确度带来了较大的偏差。传统机器学习方法gkm-SVM[6]是一种基于支持向量机的分类模型,输入DNA序列,输出该序列的打分。但是gkm-SVM只能用于小于5 000的样本数,而LS-gkm[7]在数据量方面进行了升级,改善了这一问题。最近,深度学习模型已广泛应用于该领域。例如,DeepBind[8]成功应用卷积神经网络(Convolution Neural Network,CNN)[9]来捕获主题特征,其性能优于以前的方法。DanQ[10]成功地将递归神经网络(Recurrent Neural Network,RNN)[11-12]附加到基于CNN的模型,以便可以捕获核苷酸碱基的长程相互作用[13]。但是,目前所有现有的基于机器学习的方法都在数据集上分别训练多个模型对应不同的TF和实验环境CELL,也就是不包括上下文限制。此外,这些方法也缺少对自身提取的特征值进行生物意义上的解析,进而影响对预测问题的深入研究。
本文提出一种结合深度学习和传统机器学习方法的模型semanticSVC,预测TF与DNA序列的结合特异性。首先,对来自ENCODE项目[14]的ChIP-seq数据集进行整合,使用以EMB-CNN-RNN-MLP为框架的深度学习模型进行训练,获取具有多TF全局语义的序列特征向量,本文称作semanticFeature。然后以semantic-Feature作为DNA序列特征,通过支持向量分类机(Support Vector Classify,SVC)进行目标训练,其最终预测结果AUC(Area Under roc Curve)分布要优于其他先进的方法。此外,通过对semanticFeature的降维可视化处理,不仅解析了其具有的生化意义,而且证明了本文方法应用的广泛性。
1 实验方法
本文的semanticSVC将DNA序列作为输入,并根据TF、实验环境CELL信息,即TF所在的细胞环境,选择相应的SVC模型进行特异性预测。结构如图1所示。
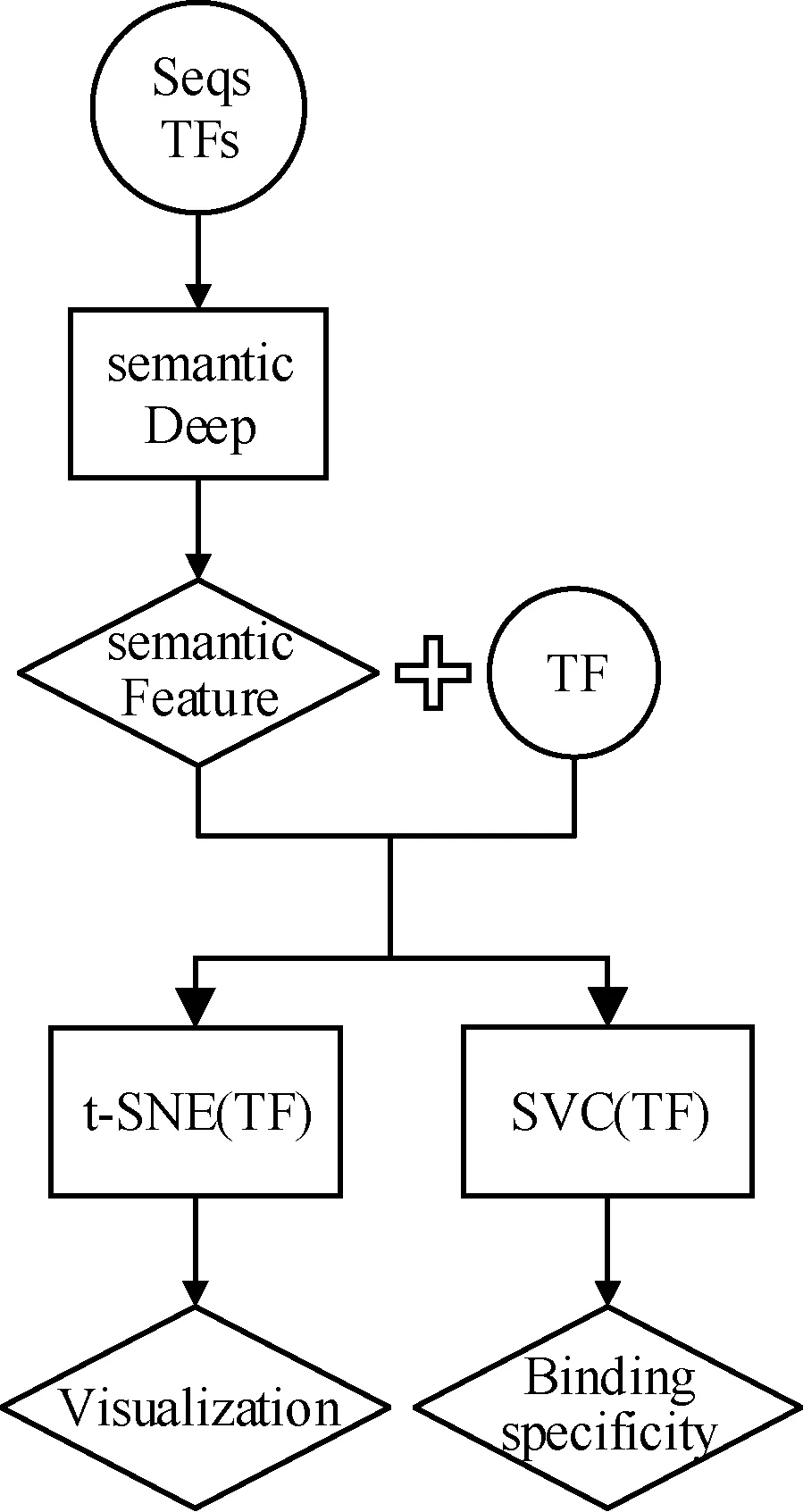
图1 semanticSVC结构
semanticSVC主要包括两部分,第一部分是基于CNN-RNN深度框架的多TF统一模型,名为semanticDeep,该模型可以有效地对DNA序列进行建模,不仅可以获取序列的长程依赖关系,而且可以解读与多个TF相关的特异性信息,这种特异性信息称作“语义特征向量”,即semanticFeature。第二部分根据不同TF,分别对基于semanticFeature的SVC模型进行构建,最终预测出DNA与TF的结合性。我们将semanticFeature进行可视化,可以直观地帮助读者进行语义分析。
1.1 数据来源
本文数据均来自ENCODE(ENCyclopedia Of DNA Elements)项目[14](https://www.encodeproject.org)的608个不同的测定实验获取的数据,测定实验主要根据TF和实验环境CELL进行分类。为了验证semantic-Feature的优越性,ENCODE项目数据被分为两部分:506个用于semanticDeep模型的构建,取名为D506;102个用于SVC模型的构建,取名为D102。这两部分的实验数据都有各自的训练集和测试集。
每个数据集包含n条DNA序列和其对应的标签,标签为1表示该序列与TF可以结合,也就是正样本;标签是0表示该序列与TF不能结合,即负样本。序列长度为101,其中正样本是通过ChIP-seq数据的峰值测量获得,而负样本序列通过采用dinucleotide shuffling经典方法[8]模拟生成。
1.2 semanticSVC模型设计
本文的semanticDeep根据数据集D506进行构建。如图2所示,模型的输入是DNA序列的词向量和TF的one-hot编码。如果把一条DNA序列看作一个句子,那么DNA序列与TF的结合位点,即motif,可以看作句子的一个单词,一个motif的长度一般是8,所以这里选取8作为词向量的k-mer[15]。输出是DNA与TF特异性的两种分类结果:0或1。对于一个序列s、TF和CELL,semanticDeep可以抽象为四个阶段,该过程表示为:
f(s,tf,cell)=
MLP(RNN(CNN(EMB(s))),(tf,cell))
(1)
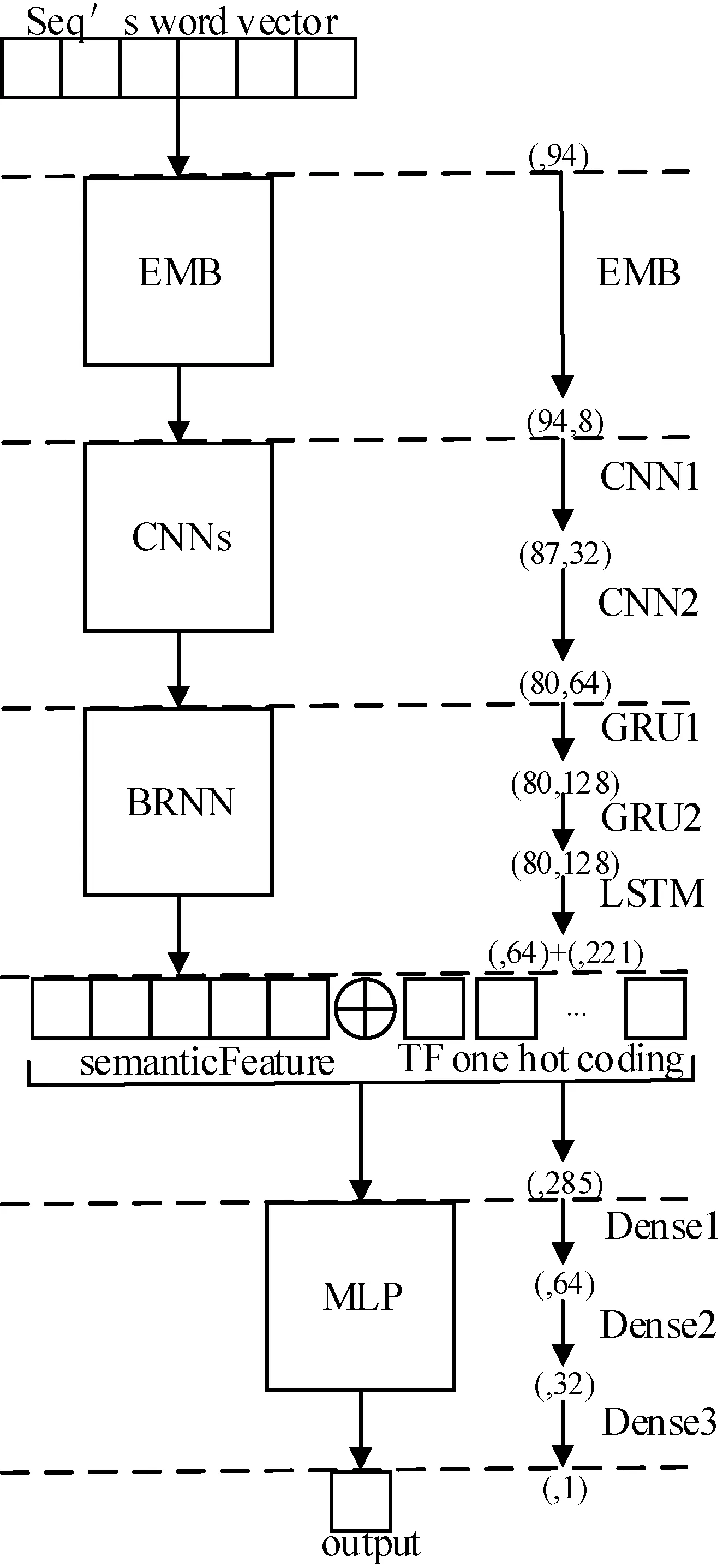
图2 semanticDeep结构
模型第一层是EMB层(Embedding)。经过EMB层,把每个序列替换为向量的索引,在训练过程中,向量会得到更新,通过索引寻找就更加快捷方便。
第二层是CNNs层。其由两层1维CNN组成,第一层CNN有32个卷积核,第二层CNN有64个卷积核。如果把碱基比作字母,那么一条DNA序列就是一条没有分词的句子,CNN可以有效地将句子分成词。每层卷积神经网络后又加了一层BatchNormalization[16],提高网络的泛化能力,避免梯度下降和梯度爆炸。
第三层是BRNN层。该层由三层RNN包括一层双向LSTM和两层双向门循环单元(Gated Recurrent Unit,GRU)构成。
LSTM中设计了输入门(input gate)、忘记门(forget gate)和输出门(output gate)对信息进行长期处理,以获得信息之间的长程依赖关系。LSTM模型的计算公式如下:
it=σ(Wixt+Uiht-1)
(2)
ft=σ(Wfxt+Ufht-1)
(3)
ot=σ(Woxt+Uoht-1)
(4)
(5)
(6)
ht=ot×tanhct
(7)
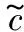
GRU的设计是将LSTM中的input gate和forget gate更改为更新门(update gate),将output gate更改为重置门(reset gate)。GRU模型的计算公式如下:
zt=σ(Wzxt+Uzht-1)
(8)
rt=σ(Wrxt+Urht-1)
(9)
(10)
ht=(1-zt)×ht+zt×ht-1
(11)
式中:z表示update gate;r表示reset gate。
由于双向GRU比双向LSTM少一个门,所以参数更少、需要的存储空间更少、训练时间更少;而双向LSTM在数据集较大时的性能更好。在双向LSTM后面加一层双向GRU是为了减少模型参数、降低模型的过拟合。最后,经过综合考虑训练性能和速度,semanticDeep采用了双向LSTM和双向GRU混用的方式。DNA序列上的一个碱基与其他碱基是息息相关的,也就是这个基本单元不仅与它前面的基本单元相关,也与它后面基本单元相关。因此使用BRNN可以建模序列的双向关系,即获取CNNs层导出的分词之间的相关性。
第四层由多层感知机(Multi-Layer Perception,MLP)构成,映射一组输入向量到一组输出向量。每一层全连接到下一层,除了输入节点,每个节点都带有非线性激活函数。为了避免出现过拟合现象,我们还在MLP之间加入了Dropout[17]层,最后使用Sigmoid函数将输出变成一个0至1之间的值。
对于D102数据集,通过semanticDeep的第三层BRNN层将DNA序列转化为具有语义的64维向量semanticFeature,最后将semanticFeature和其对应的序列标签作为本文SVC模型的输入进行训练。经训练,每个TF和CELL对应一个SVC模型,每个模型可以预测输出基于semanticFeature的DNA与该TF的结合特异性。通过SVC,可以获得数据集中的支持向量(Support Vector,SV),支持向量意味着这些DNA样例的预测可靠性不高。
对于semanticDeep模型训练,本文使用Keras作为框架,TensorFlow[18]作为后台,NVIDIA GeForce GTX 1080Ti的GPU作为计算单元,SVC模型使用sklearn.svm[19]包实现。
1.3 semanticFeature可视化
为了更好更直观地探索semanticFeature的可解释性和生物意义, 通过降维可视化的方法,对它们的分布和位置变化进行语义分析。因为64维的向量无法直观地体现semanticFeature的语义信息,为了更好地对semanticFeature在2维平面可视化,本文使用t-SNE降维工具[20]将64维的semanticFeature降至2维,其中每个2维向量代表一条DNA序列。然后将这些2维向量以坐标的形式映射到2-D图上,因此每个TF都可以形成一个散点图,图中的每个点都代表了对应序列的2维向量。根据序列的标签将非支持向量的点分成两种形状,其中:三角形的点代表不结合的序列;Ⅹ型的点代表结合的序列;支持向量的点则用普通的圆点表示。该图可以直观地展现本文方法提取出的semanticFeature的语义信息,即可以将序列分成是否结合特定TF的相反语义信息类;此外,DNA序列之间存在的相关性信息,特别是当序列发生突变时所引起的轨迹变化也可以直观展现。
2 实验与结果分析
2.1 评价指标
本文性能分析使用的评价指标是一种二分类模型的评价指标:AUC指标。
(12)
式中:positives表示正样本集;M是正样本的数目;N是负样本的数目;将样本的score值按照从小到大的顺序排序,最小的score对应了样本的rank1,以此类推得到ranki[2]。
2.2 性能分析
如图3所示是semanticSVC和Deepbind、DanQ的AUC分布。
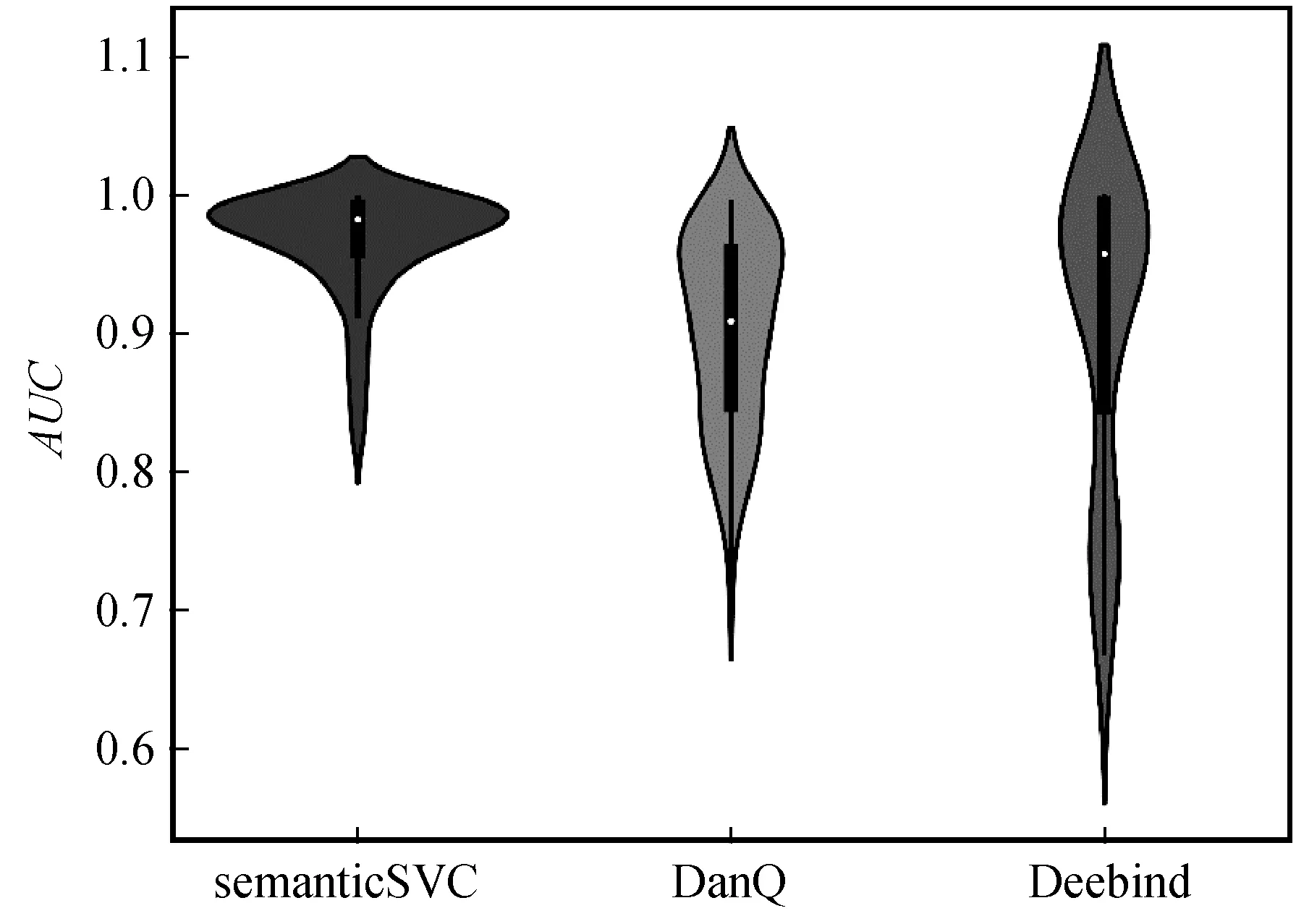
图3 三种方法的AUC分布
统计D102数据集上SVC模型测试数据集的AUC分布,发现Deepbind方法的AUC分布在0.69~0.99之间,平均分数为0.94,DanQ方法的AUC分布在0.71~0.99之间,平均分数为0.90,而本文的semanticSVC方法的AUC分布在0.82~0.99之间,平均分数达到0.96,平均准确率达到0.92。尽管三个模型的AUC最高都接近于1.00,但是从图3可以看出,本文semanticSVC方法的AUC分布比其他两个模型更加集中,这说明本文模型的训练结果不仅优于其他两个模型,而且预测准确度更加稳定。此外,也验证了基于深度学习方法的semanticDeep抽取出的semanticFeature的优越性。
接下来从数据集D102中随机选择5个测试集的AUC结果进行比较。结果如表1所示。
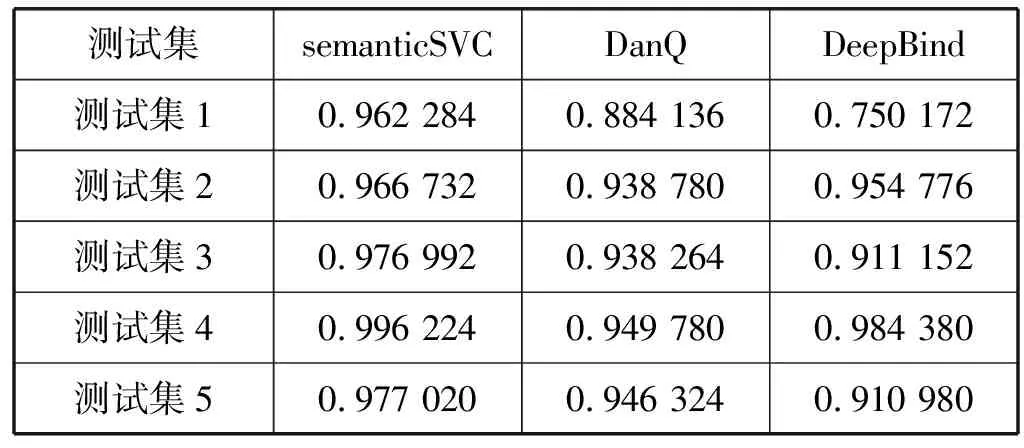
表1 5个测试数据集性能对比
各个测试数据集的信息描述见表2,测试集所在环境指的是测试集对应的TF和CELL。
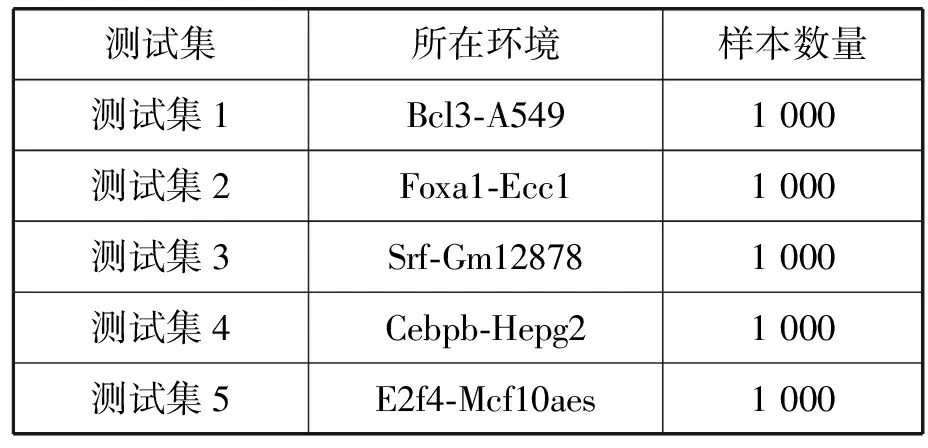
表2 5个测试数据集信息所在环境
在测试集1上DeepBind方法的AUC在0.85以下,DanQ方法的AUC在0.9以下,而本文方法AUC达到了0.96;在测试集3和测试集5上,DeepBind方法和DanQ方法均没有达到0.95,而本文方法达到了0.97;在其余两个测试集上,三个方法的AUC差距不大,均接近于1,可见本文方法在一定程度上优于其他两个方法。
2.3 semanticFeature的可视化分析
图4列举了ENCODE项目中的ChIP-seq数据对应的TF SP1 CELL K562和TF E2F1 CELL HeLa-S3的semanticFeature的散点图,其中颜色最浅的Ⅹ型点代表序列标签是1的不属于支持向量的序列,颜色较深的三角形点代表序列标签是0的不属于支持向量的序列,颜色最深的中间的圆点代表属于支持向量的序列。可以清晰地看到,属于SV的序列几乎把其他所有的序列都正确地分成两部分,左半边代表可以与TF结合的序列,右半部分代表不与TF结合的序列。已知在二分类问题的SVC算法中,SV可以决定出分割超平面的位置,也就是分类的决策边界。而属于SV的DNA序列的semanticFeature也恰好将正负样本分割在它们的两侧。同时,其他100个TF对应的ChIP-seq数据的semanticFeature的散点图也具有同样规律。
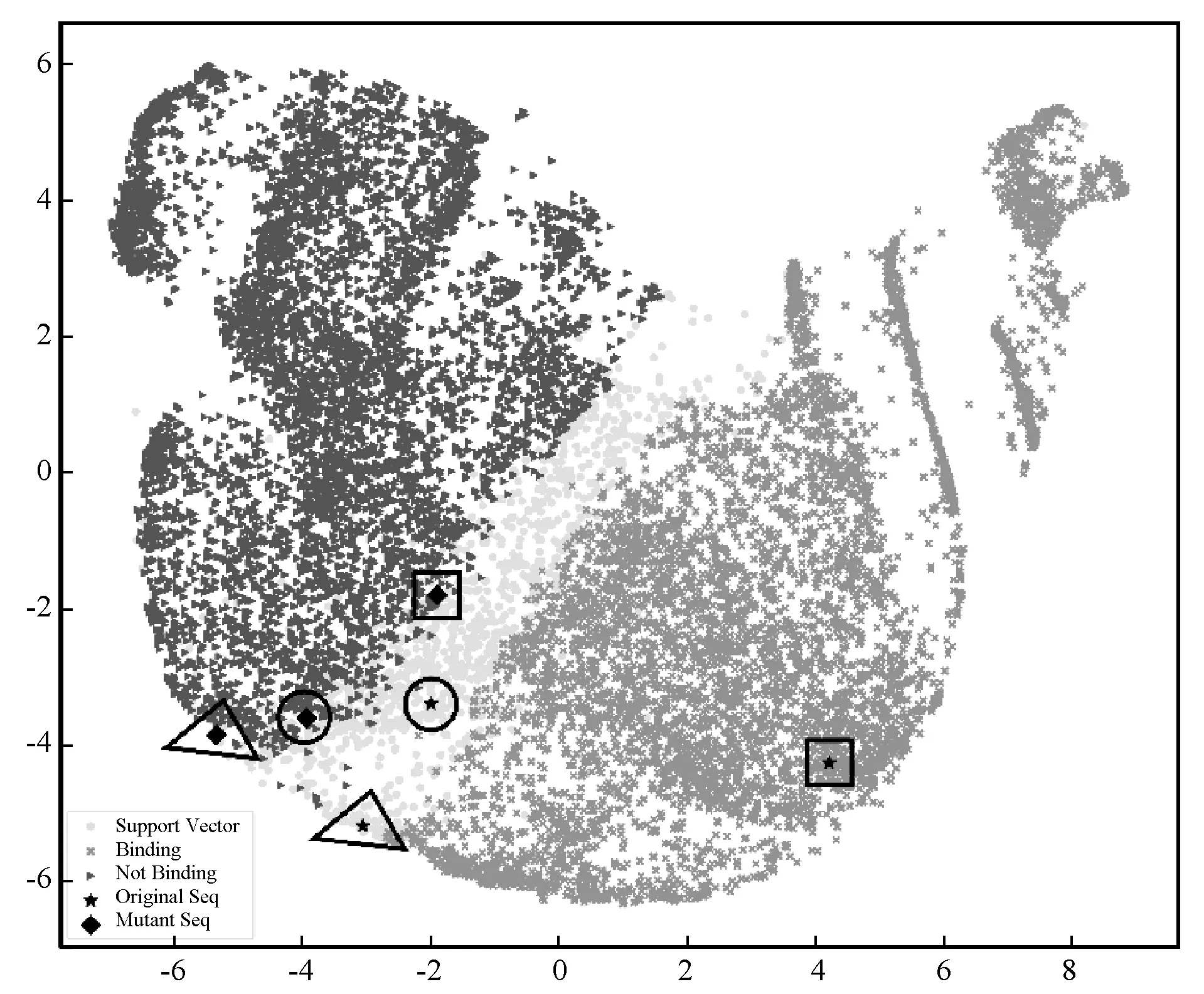
(a) TF SP1 CELL K562
(b) TF E2F1 CELL HeLa-S3图4 semanticFeature可视化
因此,可以得到这样的结论,通过semanticDeep获得的semanticFeature可以抽取到与DNA特异性相关却与TF无关的重要元级属性。从而也解释了为什么semanticSVC在仅仅使用了浅层学习方法的前提下,其预测结果却优于其他基于深度学习框架的预测器。
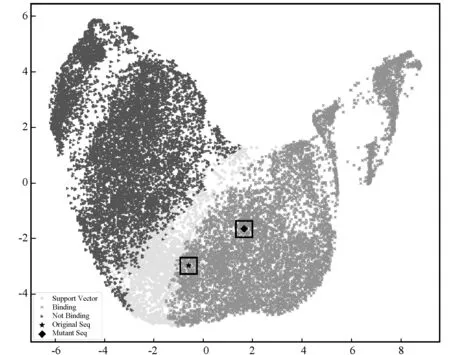
2.4 单点突变应用分析
为了验证本文方法对于单点突变的敏感性,选取来自HGMD(The Human Gene Mutation Database)[21]的两个基因HNF4A和BRCA1的四个突变序列对(Accession Number:CR1211032,CR133305,CR103902和CR090197)作为实验数据,DNA序列长度均为61,突变点位于序列中间。如图4所示,“★”代表原始DNA序列, “◆”代表突变DNA序列,矩形框里包含的是CR1211032(图4(a))和CR090197(图4(b)),圆框中包含的CR133305,三角框中包含的CR103902。其中:CR1211032、CR133305、CR103902对其基因表达的减少和功能水平的降低具有影响[22-24],而CR090197增加了启动因子的活性从而增强了与TF的结合性[25]。根据TRANSFAC database[26]存储的实验数据,分别选择与这三个结合位点有相互作用的TF SP1 CELL K562和TF E2F1 CELL HeLa-S3所对应的模型进行DNA特异性预测。根据semanticSVC预测可知,突变CR1211032(结合概率由0.99变为0.14)、CR133305(结合概率由0.55变为0.10)、CR103902(结合概率由0.57变为0.18)发生之后,其DNA序列与TF SP1的结合变低了,而突变CR076712(结合概率由0.88变为0.99)发生之后,该DNA与TF E2F1的结合变强了,以上预测结果都印证文献[22-25]中的实验结果。除此之外,通过本文方法的semanticDeep抽取出突变序列对的semanticFeature,也正确地将突变前后的变化通过二维图展现出来,例如,图4(a)中,基因HNF4A的CR1211032突变对中的原始DNA序列所在的位置处于结合区域,而突变后的DNA序列位于非结合区域,因此,我们可以推断出本文方法得到的语义特征向量semanticFeature,虽与特定的单个TF无关,但蕴涵了所有参与训练的多个TF的有用信息,从而可以直观地反映序列突变导致特异性变化的潜在因素。
3 结 语
基于生物序列建模是计算生物学的一个基础性难题,本文针对前人工作的不足之处,提出一个深度学习与浅层学习模型结合的方法:基于深度模型的语义特征提取和基于语义特征的SVC模型构建。通过可视化分析可得:对给定的DNA序列生成的语义特征向量,与特定单个TF无关,但蕴涵了所有参与训练的TF的有用信息。与现有方法相比,基于语义特征的SVC模型在预测ENCODE项目中的ChIP-seq数据的结合特异性方面取得了最优的结果。实验结果不仅证明了本文方法的有效性,而且展示了浅层机器学习模型可以通过semanticFeature快速训练其他任务。在未来的工作中我们将在序列模型中引入无监督训练机制,以使模型达到更好的泛化能力。此外我们也将尝试引入并行以及超参数优化技术来加速模型具体超参数的搜索。我们工作的最终目的是希望设计出更加符合生物意义的序列模型,从而促进计算生物学相关问题的解决。
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
健康体检与管理(2022年4期)2022-05-13
新高考·高一数学(2022年3期)2022-04-28
中国实用医药(2016年9期)2016-05-17
高中生学习·高三版(2016年9期)2016-05-14
戏剧之家(2016年1期)2016-02-25
新高考·高二数学(2015年11期)2015-12-23
长江学术(2015年1期)2015-02-27
