
基于3⁃乙基⁃1⁃(2⁃噻吩基)咪唑鎓的环金属钌配合物的合成及其对Hg2+的识别
2022-02-17 07:37李襄宏张丙广唐定国
无机化学学报 2022年2期
徐 策 杜 康 谭 琳 李襄宏 张丙广 唐定国
(中南民族大学化学与材料科学学院,武汉 430073)
环金属钌配合物是一个或多个碳阴离子作为电子给体与钌(Ⅱ)配位而形成的配合物[1-2]。与传统的Ru(bpy)32+(bpy=2,2′-联吡啶)类基于多个氮电子给体配位的钌配合物相比,碳阴离子与钌配位键的形成使得环金属钌配合物在可见光区具有更为优良的MLCT态(金属到配体的电荷转移跃迁)吸收[2-4]。此外,因环金属钌配合物既含有碳阴离子配体,又含有吡啶类配体,其结构更加灵活多变,光物理化学性质也更加丰富,已被应用于染料敏化太阳能电池中的染料敏化剂[5-8]、近红外电致变色材料[9-10]以及PDT(光动力学治疗)光敏剂[11-12]等领域,展现出了良好的应用前景。
另一方面,该配合物配体结构的灵活性使得对其修饰并引入特定离子的识别单元相对容易,且其在可见光区的光吸收变化更易于实现可视化,环金属钌配合物在小分子离子的识别领域也逐渐受到了关注[13-15]。我们以3-乙基-1-(2-噻吩基)咪唑鎓(L)为碳阴离子配体,bpy为N^N配体,合成了一种新的环金属钌配合物[Ru(L)(bpy)2]+(1),如Scheme 1所示。由于其配体中含有噻吩官能团,可与Hg2+发生作用,将该配合物用于Hg2+的识别,取得了较为满意的结果。
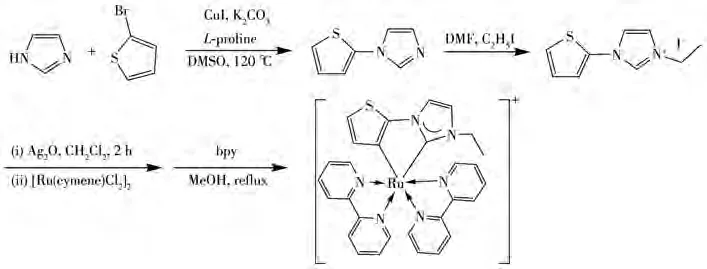
Scheme 1 Synthetic route of complex 1
1 实验部分
1.1 试剂和仪器
乙醇、乙腈、甲醇、乙酸乙酯、二氯甲烷、咪唑、无水硫酸钠、碳酸钾、二甲亚砜(DMSO)、N,N-二甲基甲酰胺(DMF)为分析纯,均购自国药集团化学试剂有限公司;碘化亚铜、L-脯氨酸、碘乙烷、bpy、2-溴噻吩为分析纯,购自阿拉丁;[Ru(cymene)Cl2]2、Ag2O为分析纯,购自北京百灵威科技有限公司。
核磁共振谱在AVAVCE-600或400(Bruker)核磁共振仪上室温测定;质谱则是在Bruker autoflex基质辅助激光解析飞行时间质谱仪(CHCA为基质)和Themo Fisher组合式高分辨液质联用仪上测定。UV-Vis吸收光谱在Shimadzu UV-2550 UV/Vis型紫外分光光度计上测定;pH值在上海雷磁pHS-3C上测定。
1.2 配体的合成
3H-1-(2-噻吩基)咪唑的合成:按文献方法[16],将CuI(1.52 g,8 mmol)、L-脯氨酸(0.93 g,16 mmol)、咪唑(2.13 g,32 mmol)、2-溴噻吩(3.87 mL,40 mmol)、碳酸钾(11.85 g,86 mmol)依次加入100 mL三口瓶中,再加入50 mL DMSO溶液。120℃搅拌16 h后,冷却至室温,减压过滤。乙酸乙酯萃取后水洗3次,无水Na2SO4干燥。旋干溶剂,柱层析得到淡黄色油状产物 0.78 g,产率 65%。1H NMR(600 MHz,CDCl3):δ 7.77(d,J=1.2 Hz,1H),7.20(t,J=1.3 Hz,1H),7.20~7.13(m,2H),7.03~6.97(m,2H)。
碘化3-乙基-1-(2-噻吩基)咪唑的合成:将3H-1-(2-噻吩基)咪唑(240 mg,1.6 mmol)、碘乙烷(0.13 mL,1.6 mmol)和20 mL DMF加入反应瓶中。通氩气10 min后,封闭瓶口,100℃下反应24 h。然后将反应液倒入乙酸乙酯中,过滤得到淡褐色固体0.45 g,产率为 92%。1H NMR(400 MHz,DMSO-d6):δ 9.77(s,1H),8.24(t,J=1.8 Hz,1H),8.07(t,J=1.6 Hz,1H),7.69(dd,J=5.4,1.4 Hz,1H),7.57(dd,J=3.8,1.4 Hz,1H),7.19(dd,J=5.4,3.8 Hz,1H),4.27(q,J=7.3 Hz,2H),1.50(t,J=7.3 Hz,3H)。
1.3 配合物1的合成
通氩气5 min后,将碘化3-乙基-1-(2-噻吩基)咪唑(120 mg,0.4 mmol)溶于25 mL无水二氯甲烷,加入Ag2O(50 mg,0.2 mmol)。常温下搅拌2 h后加入[Ru(cycme)Cl2]2(121 mg,0.2 mmol)。继续反应24 h后过滤并将滤液浓缩,氧化铝快速柱层析得到棕色固体。少量甲醇溶解后,加入有bpy(0.25 g,1.57 mmol)的三口瓶中,再加10 mL甲醇,回流12 h后停止加热。旋转蒸发至干,硅胶柱层析,展开剂为CH2Cl2/CH3OH(12∶1,V/V),得到黑色固体 72 mg,产率为19%。1H NMR(600 MHz,CD3CN):δ 8.41(d,J=8.2 Hz,1H),8.34(d,J=8.2 Hz,1H),8.34(d,J=8.2 Hz,1H),8.28(d,J=8.0 Hz,1H),8.11(d,J=5.7 Hz,1H),8.01(d,J=6.2 Hz,1H),7.95(d,J=5.5 Hz,1H),7.91~7.86(m,2H),7.84(td,J=8.1,1.5 Hz,1H),7.77(td,J=8.1,1.4 Hz,1H),7.60(d,J=5.5 Hz,1H),7.58(d,J=2.0 Hz,1H),7.34~7.27(m,2H),7.27~7.19(m,2H),7.03(d,J=2.0 Hz,1H),6.92(d,J=4.8 Hz,1H),6.04(d,J=4.8 Hz,1H),3.38(m,2H),0.78(t,J=7.2 Hz,3H)。13C NMR(101 MHz,CD3CN):δ 192.15,165.65,157.40,156.76,155.97,155.81,154.65,154.27,149.31,148.47,135.30,135.21,134.04,133.18,132.66,126.44,126.20,126.11,125.82,123.19,123.04,122.56,120.35,117.29,116.29,43.78,16.43。ESIHRMS:[M+](C29H25N6SRu)+m/z:理论值591.090 5,实验值591.090 2。
1.4 溶液的配制及吸收光谱测定
将配合物1溶解于乙腈制备储备液,然后分别用 HEPES缓冲液(pH=1.98~12.11)稀释到 20 µmol·L−1(V乙腈∶V缓冲液=2∶1)。这里HEPES缓冲溶液的不同pH值是通过向HEPES溶液中加入盐酸或NaOH溶液后由pH计测得。配合物1在不同pH值溶液中的稳定性是配好溶液后立刻在分光光度计上测试。Hg2+响应的pH范围测定是在配合物1的HEPES缓冲液(pH=1.98~12.11)中不加或加入Hg2+后分别放置30 min后测定;Hg2+滴定和其他常见金属离子干扰实验则是加入相应浓度的离子并搅拌均匀,放置3 min后测定。Hg2+溶液由Hg(NO3)2配制得到,因有毒,使用时应戴手套并特别小心。
检测限LOD测算方法如下:测定10次配合物1溶液的吸收光谱,计算546 nm处的吸光度A546nm的标准偏差σ。根据滴定曲线拟合得到配合物1与Hg2+浓度的线性相关曲线,获得其斜率k值,即可根据公式LOD=3σ/k计算得到配合物对Hg2+的检测限(3是99%置信度下的因子)。
1.5 理论计算方法
本工作中所有的计算由成都测试狗科研服务有限公司完成。采用ORCA 4.0.1软件包,计算级别是在PBE0杂化泛函方法结合D3BJ色散校正,以及def2-SVP基组下进行。Multiwfn软件用于获得Hirshfeld电荷,分子轨道图则采用VESTA显示。
2 结果与讨论
2.1 配合物1对酸碱的稳定性及Hg2+识别pH范围的选择
在合成环金属钌配合物的过程中,非常重要的一个步骤是使用碱性物质脱去芳环上的H,从而获得碳阴离子与钌(Ⅱ)进行配位[1-2]。基于此,环金属钌配合物中的Ru—C键在酸性介质中极有可能受到影响,从而导致光谱发生变化[1,17-18]。因此,我们研究了配合物1对酸碱的稳定性。首先,我们测定了配合物1和碘化3-乙基-1-(2-噻吩基)咪唑的UV-Vis吸收光谱,结果如图1所示。位于245 nm处的吸收峰可归属于碘化3−乙基−1−(2−噻吩)−咪唑的π→π*跃迁吸收。而属于2,2′-二联吡啶配体的π→π*跃迁强吸收则位于295 nm。出现在350 nm处的较强吸收应归属于配体到金属的电荷转移跃迁吸收,即LMCT吸收。450~750 nm范围出现的中等强度吸收带是由金属到配体的电荷转移跃迁(MLCT)吸收引起。这里有2个明显的吸收峰,前者位于487 nm,后者位于546 nm。根据其吸收峰位及摩尔消光吸收系数值,这2个吸收峰分别可归属于金属钌(Ⅱ)到碳阴离子配体的电荷转移跃迁吸收,以及金属钌到联吡啶配体的电荷转移跃迁吸收[3-4]。之后,我们考察了配合物1在不同pH值的溶液中,其UV-Vis光谱在波长546 nm处的吸光度(A546nm)变化。如图2所示,在中性及碱性条件下,配合物1在546 nm处的吸光度并未发生明显变化。而在酸性条件下,吸光度有明显的下降,且随着酸度增大,吸光度下降得更显著。
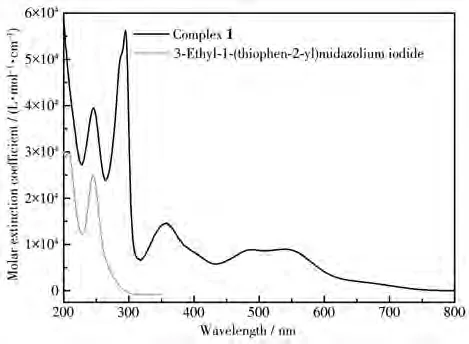
图1 配合物1和碘化3-乙基-1-(2-噻吩基)咪唑在乙腈中的UV-Vis谱图Fig.1 UV-Vis spectra of complex 1 and 3-ethyl-1-(thiophen-2-yl)-imidazolium iodide in CH3CN solution
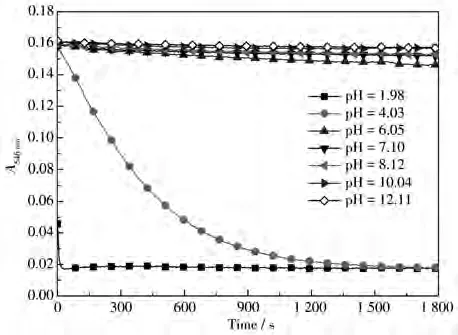
图2 在不同pH值的CH3CN/HEPES溶液中,配合物1在546 nm处的吸光度变化Fig.2 Absorbance changes at 546 nm of complex 1 in CH3CN/HEPES solutions with different pH values
配合物1含有噻吩官能团,可利用其S原子的亲汞性达到对Hg2+识别的目的[14,19]。考虑到配合物1在酸性环境中的不稳定性以及碱性环境中金属离子易水解的特点,这里着重考察了pH对配合物1与Hg2+反应的影响。为排除酸性条件下配合物中Ru—C键可能断裂而产生的影响,根据图2所得信息,将配合物1在pH=1.98~12.11的空白溶液和加入Hg2+的溶液均放置30 min后测试。如图3所示,加入Hg2+后,在pH=6.05~12.11范围内,配合物1在546 nm处的吸光度均发生明显变化,其中在pH=6.05~8.12范围内最为明显。由此可见,在pH=6.05~8.12范围内,配合物1与Hg2+之间相互作用最强。
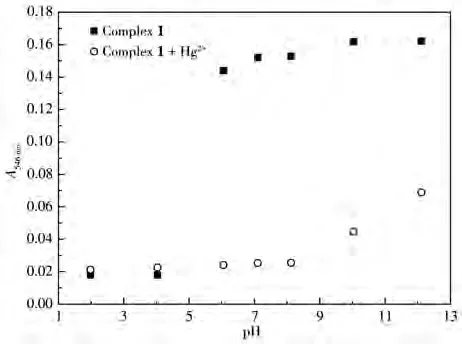
图3 配合物1在不同pH值的CH3CN/HEPES溶液中A546 nm的变化Fig.3 A546 nmchanges of complex 1 in CH3CN/HEPES solutions with different pH values
2.2 配合物在CH3CN/HEPES中对Hg2+的识别
配合物 1在 CH3CN/HEPES(2∶1,V/V,pH=6.98)溶液中与不同浓度Hg2+反应的吸收光谱变化如图4所示。随着Hg2+浓度的增加,配合物1在546 nm处的吸收峰强度逐渐减弱,并在448 nm处出现新的吸收峰,最大吸收波长从546 nm蓝移至448 nm,溶液的颜色由紫红色变为黄色。根据配合物1与Hg2+的滴定曲线获得配合物1的ΔA546nm与Hg2+浓度的线性相关曲线。如图4插图所示,当Hg2+浓度在0~12µmol·L−1范围内,二者呈现良好的线性关系,通过计算可知配合物1对Hg2+的检测限为0.057 µmol·L−1。
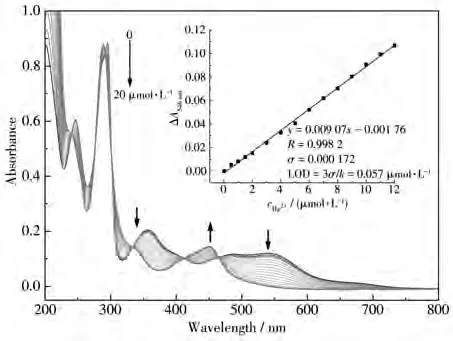
图4 随着Hg2+浓度增加,配合物1在CH3CN/HEPES中的吸收光谱变化Fig.4 Absorption spectral changes of complex 1 in CH3CN/HEPES solution with increasing concentration of Hg2+
2.3 离子干扰实验
考虑到选择性在检测分析物时的重要性,考察了配合物 1 在 CH3CN/HEPES(2∶1,V/V,pH=7.00)溶液中与各种金属离子反应的情况。如图5所示,K+、Na+、Mg2+、Ca2+、Zn2+、Cd2+、Mn2+、Pb2+、Ag+、Co2+、Ni2+、Fe2+、Cr3+、Al3+的加入并未引起明显的吸收光谱变化,A546nm/A448nm值在1.2~1.3范围内变化。向含有上述各金属离子的溶液中再加入Hg2+时,A546nm/A448nm值由1.2~1.3下降至0.2~0.3。这表明:这些金属离子共存时并不干扰配合物1对Hg2+的识别。
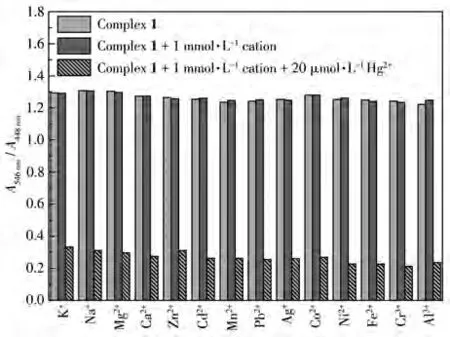
图5 配合物1在CH3CN/HEPES溶液中加入各种金属离子后的UV-Vis光谱响应Fig.5 UV-Vis spectrum responses of complex 1 upon addition of various metal ions in CH3CN/HEPES solutions
2.4 识别机理分析
图6为配合物1与Hg2+作用的Job′s plot图。由图可知,配合物1与Hg2+结合比例为1∶1,这与配合物1结构中含有一个S原子是一致的。Wu等通过DFT计算分析了含有噻吩官能团的中性铱配合物和阳离子型铱配合物的表面电荷分布,发现阳离子型铱配合物的电子云密度比中性铱配合物低,导致其与Hg2+的结合能力减弱,因而阳离子型含噻吩的环金属铱配合物很难实现对Hg2+的识别[20]。对于本工作中的环金属钌配合物1,其配体3-乙基-1-(2-噻吩基)咪唑鎓是离子型的,然而经过DFT计算发现:其与Ru(Ⅱ)配位后,HOMO轨道主要分布在环金属配体和联吡啶及钌金属中心,LUMO轨道则离域在2个联吡啶配体上(表1)。原子电荷分布如图7所示,S上的电荷为0.061,比文献所报道的中性铱配合物中S原子上的表面电荷低得多[20],这说明噻吩S上仍具有较高的电子密度,因此在与Hg2+作用时,仍然可与Hg2+发生配位作用,进而改变配合物配体上的电子分布,导致吸收光谱发生变化。
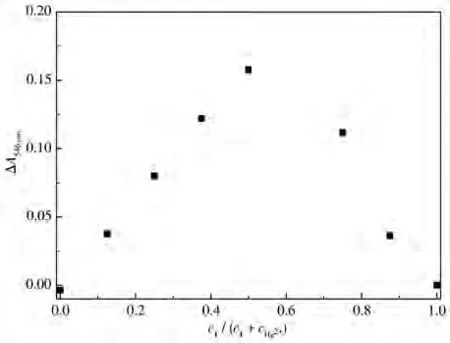
图6 配合物1与Hg2+的Job′s plot图Fig.6 Job′s plot for complex 1 and Hg2+
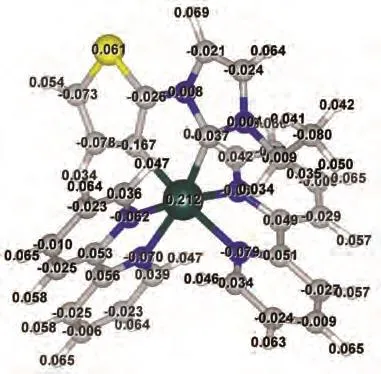
图7 配合物1的各原子电荷分布图Fig.7 Charge distributions of the atoms in complex 1
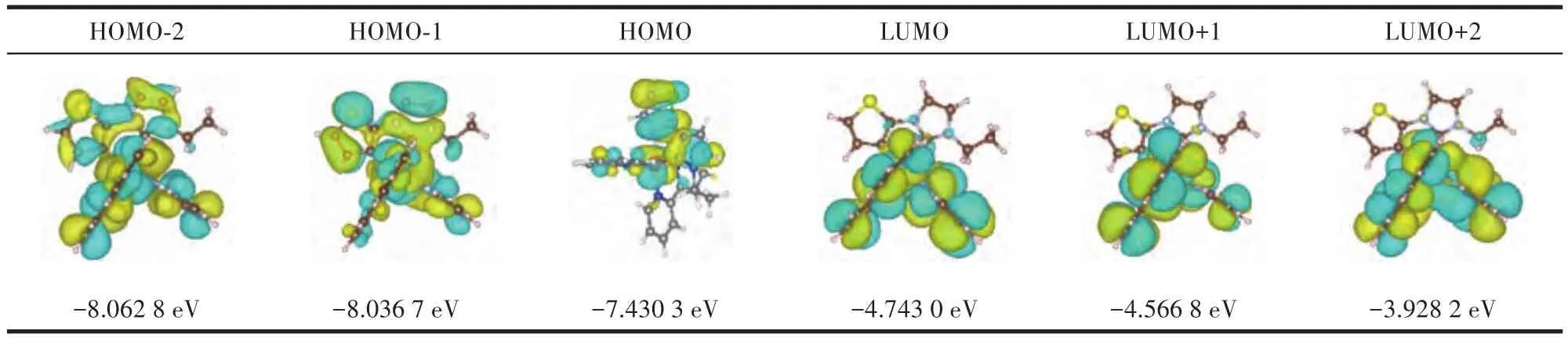
表1 配合物1的HOMO和LUMO轨道分布及能级Table 1 Molecular orbital distributions and energy levels of complex 1
值得一提的是,将配合物1置于酸溶液中和含Hg2+的中性溶液中,发现配合物1在酸溶液中的吸收光谱与配合物1在Hg2+存在时的吸收光谱几乎一致(图8),最大吸收峰均出现在448 nm,且均在410和463 nm处出现等吸收点。考虑到配合物1的溶液在加入Hg2+前后明显的颜色和吸收光谱变化,结合已报道的环钌化2-噻吩衍生物特殊的酸异构现象[21-23],我们推测该配合物在加入Hg2+后,可能使Ru—C配位向Ru—S转化。随后,将含有Hg2+的配合物1溶液(pH=6.98)进行质谱分析,发现其分子离子碎片峰出现在591.50,与异构后的分子离子碎片峰[M−H+]+计算值591.10接近(图8插图)。考虑到配合物1((C29H25N6SRu)+,m/z=591.09)的分子离子峰M+碎片也位于此处,并结合异构后所形成的配离子(C29H26N6SRu)2+带2个正电荷的结构特点,将配合物1 在 pH=2.02(不含 Hg2+)和 pH=6.98(含有 Hg2+)的CH3CN/HEPES(2∶1,V/V)溶液中分别加入NaCl,充分搅拌后离心,将所得溶液分别进行质谱分析,得到如图S1(Supporting information)所示的高分辨质谱。从图中可以看到,在酸性溶液和含Hg2+的溶液中,均出现归属于(C29H26N6SRu)2+结合 Cl−所得的[M+Cl]+分子离子峰(理论值:627.067 2),分别位于627.064 1和627.064 2。这一分子离子峰明显不同于配合物1,进一步证实了在Hg2+存在的溶液中,形成了Ru—S配位模式的配合物。根据以上数据分析,推测配合物1与Hg2+可能的作用机理如Scheme 2所示。
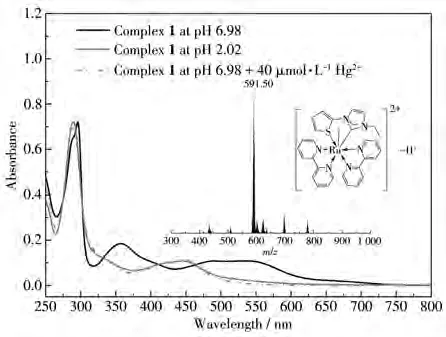
图8 配合物1在CH3CN/HEPES溶液中不同条件下的UV-Vis谱图Fig.8 UV-Vis spectra of complex 1 in CH3CN/HEPES solutions under different conditions
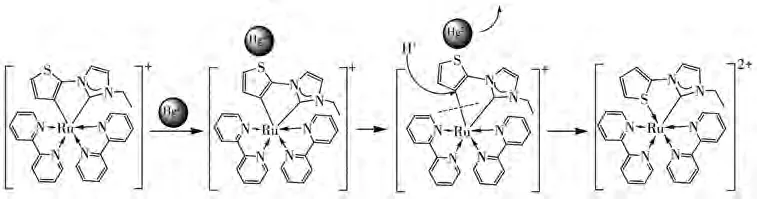
Scheme 2 Possible mechanism of complex 1 recognizing Hg2+
3 结论
合成了基于3-乙基-1-(2-噻吩基)咪唑鎓的环金属钌配合物1,探究了常见金属离子对其吸收光谱的影响。结果表明仅Hg2+能够引起配合物1的吸收光谱发生100 nm蓝移,这可能是Hg2+与噻吩硫作用引起配位异构化反应所致。经计算,配合物1对Hg2+的检测限为0.057 µmol·L−1。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
武汉工程大学学报(2022年4期)2022-08-26
昆钢科技(2022年2期)2022-07-08
医学概论(2022年4期)2022-04-24
世界农药(2021年11期)2021-12-09
昆钢科技(2021年4期)2021-11-06
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
鞍钢技术(2020年5期)2020-10-10
分析化学(2018年12期)2018-01-22
分析化学(2017年12期)2017-12-25
山东工业技术(2016年15期)2016-12-01
