
Clinical manifestations and prenatal diagnosis of Ullrich congenital muscular dystrophy: A case report
2022-02-11 05:29JunHuYanHuiChenXinFangYuZhouFengChen
World Journal of Clinical Cases 2022年1期
INTRODUCTION
Ullrich congenital muscular dystrophy (UCMD, OMIM: 254090) is one of the collagen-VI-related myopathies caused by mutations of the,, andgenes. The prevalence of UCMD is not sufficiently known, with an estimated 0.13 per 100000 in Northern England[1], which is higher than that reported in China[2]. Affected individuals are characterized by muscle weakness, proximal joint contracture, distal joint hyperlaxity, and progressive respiratory failure[3,4]. Given that the disease may severely affect quality of life and lifespan and that no curative care is available to date, genetic counseling and prenatal diagnosis are crucial for the families at risk.
Here, we report the clinical manifestations and prenatal diagnosis of compound heterozygous mutations of thegene in a Chinese family with UCMD. A third UCMD child of the family was prevented from birth through prenatal diagnosis. The study protocol was carried out in compliance with the Declaration of Helsinki and approved by the Ethics Committee of Fujian Medical University (No. 2020KY023). Written informed consent was obtained from the parents.
It s the most amazing thing that will ever happen in my life. This was Roger s first National League home run, and I caught the ball. Tears rolled down my face. Roger came running out at the end of the inning and said, I can t believe it. I said, You can t? I can t!
CASE PRESENTATION
Chief complaints
A 3-year-old boy and his 4-year-old brother presented with their disabilities in walking independently after birth.
15.Ball:A ball is a large party in which the participants dress up in their finest clothes and dance. Balls were exclusively for the privileged and wealthy.
History of present illness
Tissue sections obtained by muscle biopsy of the quadriceps femoris in the younger brother were examined after hematoxylin and eosin staining and immunostaining with anti-collagen VI antibodies. Muscle pathology showed the myofibers were of different sizes. They were atrophied, non-necrotic, or broken. There were no intracellular lipid droplets, glycogen vacuoles, bordered vacuoles, or rods in the myofibers. The endomysial connective tissue demonstrated slight hyperplasia. Histochemical examination showed that collagen VI stained positive at the sarco-lemma but negative at the endomysium and perimysium of the myofibers (Figures 2A and 2B).
Then she drew her breath, opened her eyes, and said, Ah! where am I? You are with me, dear lady, he answered, and told her all that had happened, and how he had brought her to life again
History of past illness
The patients’ parents, a 35-year-old Chinese father and a 32-year-old Vietnamese mother, were unrelated, neither of whom reported a family history of neuromuscular diseases.
Personal and family history
The brothers were delivered by cesarean section at term after a simple pregnancy. They began to hold up their heads at the age of 3 mo and could sit by themselves at 8 mo. Their language and social communication abilities were normal.
Physical examination
The brothers’ conditions gradually deteriorated. The elder brother died of respiratory failure in October 2020. The younger brother is unable to stand now. His joint contractures and scoliosis worsen than before. The parents opted after prenatal diagnosis for voluntary interruption of the third UCMD pregnancy.
Laboratory examinations
Creatine kinase levels of the brothers were normal. Basic lung tests were normal too. The IQ on the Gesell developmental diagnostic scale was 89.0 and 81.0, respectively.
Genomic DNA extracted from the peripheral blood of both the patients and their parents was analyzed by whole-exome sequencing[5]. High-throughput sequencing was performed on an Illumina NovaSeq 6000 Series Sequencer (PE150). Sanger sequencing was used to confirm the mutations. Both brothers were found to carry a heterozygous frameshift mutation c.1353_c.1354insC (p.Arg453ProfsTer42) in exon 16 of thegene inherited from their mother and a heterozygous nonsense mutation c.2105G>A (p.Trp702Ter) in exon 26 inherited from their father.
28. Crumb17: Note that just as bread provides life sustaining sustenance85, the children are now depending on it to save their lives beyond it s true purpose.
The younger brother stood and walked with assistance at the age of 15 mo. He has achieved no further motor milestones since that time. The elder brother could hardly stand, even with help, and could never walk independently.
In this study, we report a family suffering from UCMD living in China. In the clinic, patients with UCMD exhibit a wide spectrum of clinical severity with variable motor and respiratory weakness. Three subtypes of UCMD are defined according to walking status: Patients who never achieve ambulation are categorized as early-severe subtype; those who walk but lose this ability (or are about to lose it) are categorized as moderate-progressive subtype; and patients who are still fully ambulatory are categorized as mild subtype[3,4,8-10]. The brothers had the typical manifestations of the early-severe subtype: A delayed motor milestone (never walking independently), torticollis, scoliosis, proximal joint contracture, distal joint hyperextension, right hip joint dislocation, and calcaneal protuberance. Basic lung testing of the brothers was normal, which is consistent with the other reports that describe patients with the earlysevere subtype of UCMD as experiencing early respiratory failure at around 10 years old[11].
Imaging examinations
Radiography showed cervical thoracolumbar scoliosis and right hip dislocation. Brain magnetic resonance imaging was normal.
FINAL DIAGNOSIS
Early-severe subtype of UCMD.
TREATMENT
There is currently no cure for UCMD. Treatment is mainly supportive against symptoms. The brothers underwent rehabilitation therapy regularly, including muscle massage and passive exercise. Six months later, their mother was pregnant with a third child. Prenatal diagnosis was performed using DNA samples extracted from the fetal amniotic fluid at 20 wk of gestation. Multiplex ligation-dependent probe amplification (MLPA) was used to detect UCMD-related gene mutations[6]. Linkage analysis of short tandem repeats (STRs) was used to identify maternal blood contamination and the biological parents[7]. Prenatal diagnosis revealed that the fetus was positive for the same mutations (Figures 2C and 2D). After a detailed consent process, the pregnancy was terminated.
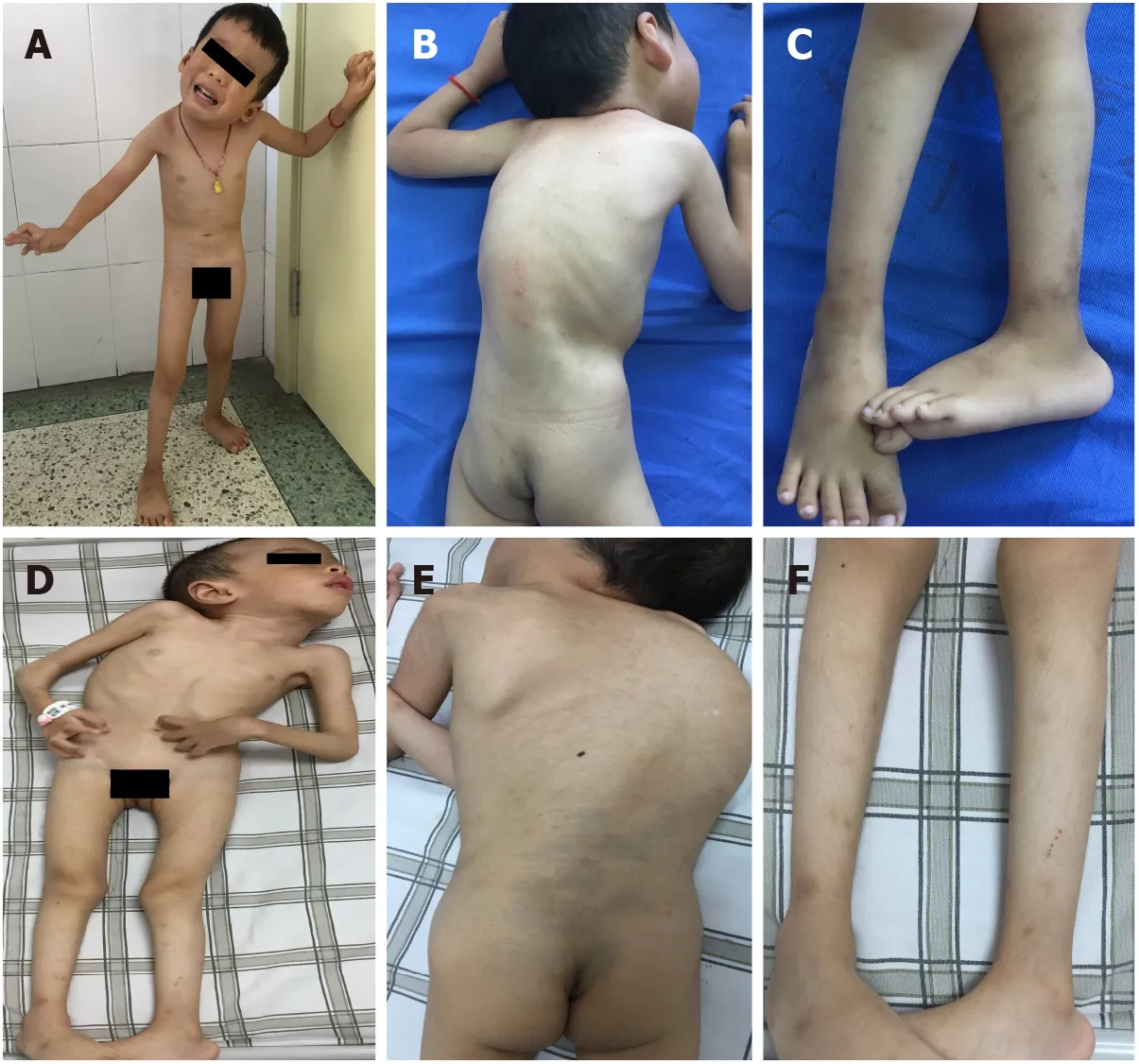
OUTCOME AND FOLLOW-UP
The younger brother had torticollis and scoliosis, could sit by himself, stand and walk with help, but could never walk independently. His muscle tone was generally decreased, and muscle power was graded as 3 or 4 in the extremities. Proximal joint contractures, such as at the shoulders, hips, and knees, were prominent bilaterally. Distal joints, including wrists, fingers, and toes, were markedly hyperextensible. Calcaneal protuberance was remarkable. Muscular atrophy was visible in the whole body. Knee reflexes were present. Pathological signs in the central nervous system were negative (Figures 1A-C). The elder brother had similar clinical manifestations, with the degree more serious than that of the younger brother (Figures 1D-F).
DISCUSSION
10. Husband: The father/husband s role in the tale is an interesting one. While the father is usually the birth father of the children, he has different levels of responsibility for the abandonment across versions of the tale. In some versions he willingly leaves the children in the forest. In other versions, he ineffectively protests their abandonment. The textual hint that the wife s wishes will win over the children s safety comes from the word choice of husband over father to describe the man s primary role.Return to place in story.
Based on the clinical features, we initially focused on the collagen VI genes, and finally identified two mutations of thegene (c.1353_c.1354insC, p.Arg-453ProfsTer42/c.2105G>A, p.Trp702Ter) in both brothers. The heterozygous variation (c.1353_c.1354insC) is a frameshift mutation, which changes amino acid synthesis starting from amino acid 453 (arginine) and ending with the 42amino acid (p.Arg453ProfsTer42). Another heterozygous variation (c.2105G>A) is a nonsense mutation, which prematurely terminates peptide synthesis by changing the codon of amino acid 702 (tryptophan) to a stop codon (p.Trp702Ter). The heterozygous mutations affected collagen VI function, which was classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines: PVS1 + PM2 + PP1 + PM3 and PVS1 + PM2 + PP1 + PP3 + PM3[12]. The brothers had compound heterozygous mutations inherited by autosomal recessive patterns[13].

Variable degrees of histological changes can be observed in muscle biopsies of patients with UCMD. The spectrum includes fiber size variation, increased endomysial connective tissue or adipose tissue, and mild necrotic and regenerating process. Collagen VI staining in muscle biopsies of patients with UCMD is variably less or full absent in the extracellular matrix. It is present in the interstitium but is absent or reduced in the sarcolemma[8]. The muscle pathology of the younger brother was consistent with myogenic damage and regarded as collagenopathy. It was in line with the pathological changes of UCMD. Therefore, we considered the brothers as having early-severe subtypes of UCMD.
UCMD seriously affects quality of life and lifespan and no curative care is available to date, which brings a heavy burden to the family and society. Genetic counseling and prenatal diagnosis are crucial for the families at risk, as the autosomal recessive genetic disease affects a quarter of the patient’s siblings. UCMD demonstrates genetic and phenotypic variability. In familial cases, the genetic background must be identified accurately for reliable counseling and prenatal diagnosis[14,15]. The brothers had the typical manifestations of early-severe subtype UCMD and carried compound heterozygous mutations of thegene. The absence of collagen VI staining in the younger brother’s muscle was identified accurately. It was appropriate to offer genetic counseling (including discussion of the potential risks to offspring and reproductive options) to the parents, who were carriers. Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing were feasible. Differences in perspectives may exist among medical professionals, and within families, regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues can be helpful.
The fact was that the Fairy of the Beech-Woods, thinking she had been punished enough, had withdrawn66 the enchantment from her, and transferred it to Featherhead, thereby67 in an instant depriving him of the good looks which had done so much towards making him the fickle31 creature he was
After their mother was pregnant with a third child, the parents requested prenatal diagnosis for the pregnancy. As the brothers had the typical manifestations of the early-severe subtype UCMD and carried compound heterozygous mutations of thegene by whole-exome sequencing, and the absence of collagen VI staining in the younger brother’s muscle was identified accurately, this allowed us to offer MLPA + STRs as a prenatal diagnostic test for this subsequent pregnancy. However, the detection of gene mutations by this approach may not be reliable due to high genetic heterogeneity and detection errors. All the risks, benefits, and limitations of the chosen testing plan were explained to the parents. We proceeded with the fully informed consent from the parents, particularly with respect to the research nature of these tests, and with ethics committee approval. MLPA + STRs confirmed that the fetus carried the same two mutations of thegene in the amniotic fluid. The parents were told that the fetus would likely suffer from UCMD after birth. They needed to decide whether to continue with the pregnancy. After a painful psychological struggle, the parents finally decided to terminate the pregnancy.
CONCLUSION
We report a Chinese family suffering from UCMD caused by compound heterozygous mutations of thegene (c.1353_c.1354insC/c.2105G>A). By clarifying the type and source of the disease-causing mutations in the probands, the parents had the opportunity to opt for voluntary interruption of the third UCMD pregnancy through prenatal diagnosis.
ACKNOWLEDGMENTS
We thank the boys and their parents who took part in this study. We are also grateful to all the experts involved in the study and support from Fujian Medical University Union Hospital.
World Journal of Clinical Cases2022年1期
- World Journal of Clinical Cases的其它文章
- Hepatitis B virus reactivation in rheumatoid arthritis
- Paradoxical role of interleukin-33/suppressor of tumorigenicity 2 in colorectal carcinogenesis: Progress and therapeutic potential
- Changes in rheumatoid arthritis under ultrasound before and after sinomenine injection
- Benefits of multidisciplinary collaborative care team-based nursing services in treating pressure injury wounds in cerebral infarction patients
- Outcomes and complications of open, laparoscopic, and hybrid giant ventral hernia repair
- Surgical resection of intradural extramedullary tumors in the atlantoaxial spine via a posterior approach