
典型宁东煤热解阶段硫迁移路径的分子动力学模拟*
2022-02-10 06:53梁文政王凤印王翠苹岳光溪
煤炭转化 2022年1期
梁文政 王凤印 王 坤 王翠苹 岳光溪,3
(1.山东科技大学土木工程与建筑学院清洁能源实验室,266590 山东青岛;2.青岛大学机电工程学院,266071 山东青岛;3.清华大学能源与动力工程系,100084 北京)
0 引 言
煤相关的热化学实验证实,煤在热解阶段会析出挥发分和绝大多数的污染物[1],煤热解特性会影响热解产物的分布和品质,同时煤热解特性也是设计相关反应器重点关注的因素之一[2],因此,对煤热解过程的研究尤为必要。研究[3-7]表明,热解温度、热解时间和热解气氛会显著影响煤热解产物的气、液相分布。为提高煤热解速率和热解产品质量,学者们多年来开展了许多关于煤催化热解及煤与生物质共热解的实验研究[8-13],并取得了多项优异的成果。
尽管通过实验研究可对不同条件下煤热解的宏观结果进行详细分析,但是对于微观层面的煤热解的研究尚少,这是由于煤热解涉及的反应过程极为复杂,主要由自由基驱动进行,难以通过实验手段进行检测与分析,因此,利用计算机辅助计算的反应力场分子动力学(Reax FF MD)方法日益兴起[14-18]。以往主要是通过构建简单的煤化学模型进行煤热解过程的机理研究,例如ZHAN et al[19]采用Hatcher次烟煤模型和Reax FF反应力场模拟解释了典型热解产物生成机理;SAL MON et al[20]模拟了Morwell褐煤模型热解过程中官能团的热解与生成规律;BHOI et al[21]对比了Wender褐煤模型在热解和燃烧过程中势能的变化及中间体的形成;CHEN et al[22]模拟了几种常见煤的简单模型化合物的加氢反应,证明了加氢反应的活性中心不仅与超离域性有关,还与空间位阻效应有关;ZHOU et al[23]研究了Wender褐煤模型及其水煤浆的热解/气化过程和引发机理,发现褐煤热解过程中桥键首先断裂,羧基、甲氧基和甲基等官能团随后分离,小的中间结构之间的反应会生成气体产物。
由于煤的结构复杂难以被准确解析,因而采用简化的煤结构模型往往造成分子动力学研究机制的不准确[24]。随着科技的进步,分析手段的精度不断提高,计算机性能不断发展,建立更精细更准确的煤化学结构成为可能。已有研究者[25-28]利用X射线光电子能谱(XPS)、13C固体核磁共振(13C-NMR)等分析手段来构建煤化学结构,通过计算模型相关物性参数并与实际测试数据进行比对,发现模拟值和测试值吻合程度良好,从而证明以上分析表征手段与模型建立过程具有有效性、合理性。这使得煤热化学转化研究得以在分子层面展开,为探究复杂的化学反应机理提供了支撑。
上述煤化学结构模型建立与机理研究中,大多只关注了C,H,O元素而没有关注S元素,因此,本研究基于上述煤化学结构分析方法,选取典型的红石湾宁东煤作为研究对象,通过元素分析、工业分析、XPS、13C-NMR等分析手段,构建含硫的宁东煤干燥无灰基的化学结构。使用LA MMPS程序包进行不同升温速率下宁东煤热解反应的分子动力学模拟研究,探究热解过程中典型产物分布特点,揭示宁东煤热解过程中硫元素的迁移行为,以期为含硫污染物的调控和燃烧中控制机理研究提供新思路。
1 实验部分
1.1 煤样处理与分析
实验前将宁东煤煤样研磨至粒径约为0.1 mm,采用元素分析仪(VARIO EL III)测定煤样中C,H,N元素的含量,采用全自动定硫仪(ZDL-9)测定煤样中S元素的含量,由差减法计算得到O元素含量。工业分析参考GB/T 212-2008中的方法进行。宁东煤的元素分析和工业分析结果见表1。
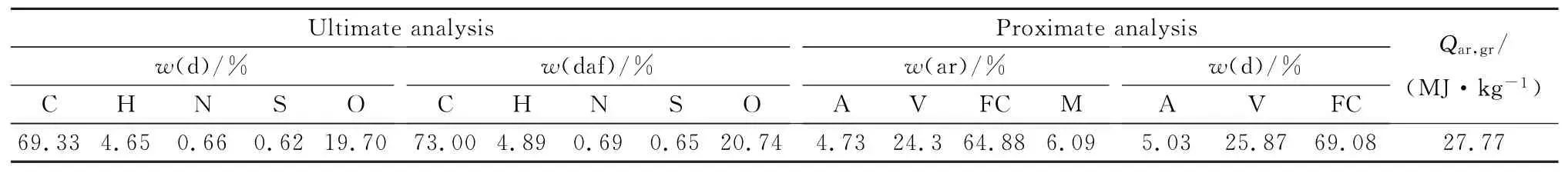
表1 宁东煤的元素分析和工业分析Table 1 Ulti mate and proxi mate analyses of Ningdong coal
通过归一化处理得到煤化学结构中各原子与C原子的个数比,结果见表2。由于S元素含量很低,为获得S元素在热解阶段的迁移路径,应保证所构建的煤化学结构中至少存在1个S原子,那么C原子应为300个,由此构建的干燥无灰基下完整的宁东煤化学式为C300H242O64N2S。
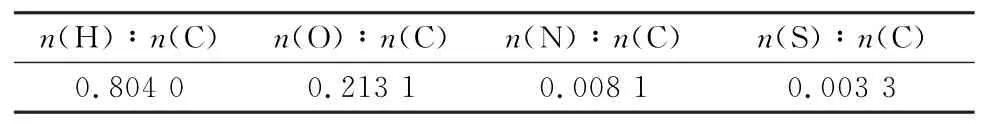
表2 宁东煤各原子与C原子的个数比Table 2 Nu mber ratio of each ato m to C ato m of Ningdong coal
1.2 煤样的物性表征与分析
煤化学结构的建立,还需确定各原子存在形式及成键方式。XPS可以用于分析物质中元素的存在形态及化学环境。对宁东煤C,O,N,S元素进行精细谱扫描并分峰拟合,得到各元素原子形态分布特征,结果见表3。
由表3可以看出,煤中C原子主要以4种形式存在,分别为以C—C键或C—H键形式存在,以代表醇、酚、醚键的C—O键形式存在,以及以代表羰基和羧基的CO键形式存在。O原子主要以3种形式存在,分别为以C—O键形式存在,以CO键形式存在,以少量金属氧化物形式存在。N原子主要以吡咯和吡啶的形式存在。S原子主要以巯基的形式存在。考虑到分子式中N原子和S原子个数较少,用吡咯和巯基分别作为N原子和S原子的存在形式。
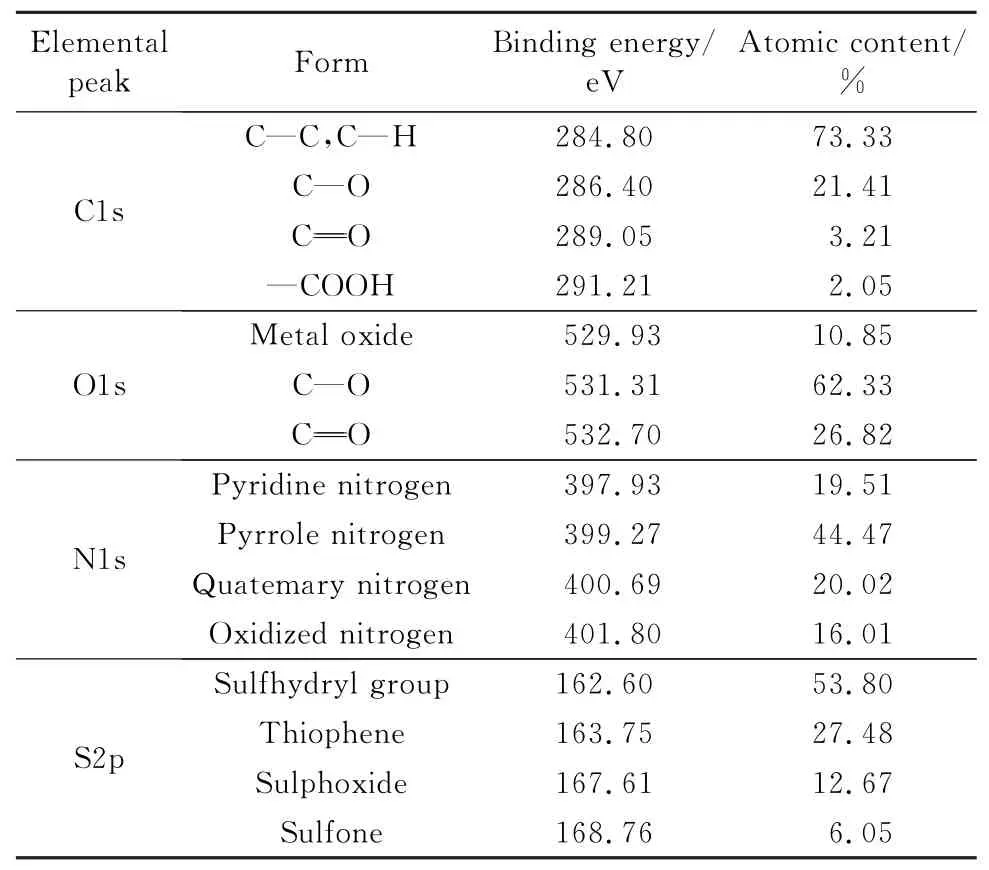
表3 宁东煤C,O,N,S原子分布特征Table 3 Distributions of C,O,N and Sfor ms of Ningdong coal by XPS analysis
13C-NMR技术可用于分析煤的平均碳骨架结构,宁东煤的13C-NMR结构参数计算结果见表4。通过公式(1)计算桥碳与周碳比XBP,从而反映煤中芳环的平均缩聚程度,其结果为0.174 1,由此得知宁东煤中结构单元以萘环和苯环为主,结合元素分析可求出各结构单元的个数[28]。
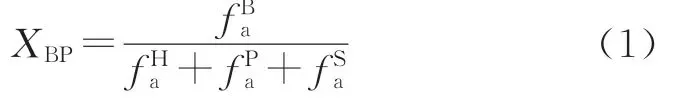

表4 宁东煤13 C-NMR结构参数计算结果Table 4 Calculation results of 13 C-NMRstructural parameters of Ningdong coal
1.3 宁东煤化学结构模型构建与优化
各结构单元间采用碳链连接构成宁东煤化学结构的基础碳骨架;根据XPS的分析结果在基础碳骨架上添加各含氧官能团、吡咯结构和巯基结构,从而建立初始的化学结构模型。一般来说,搭建的初始模型与平衡状态时的结构存在较大差别,需进行结构优化以使体系局部能量减小,从而简化计算过程[27,29]。在Material St udio软件中采用Forcite模块对所构建化学结构模型进行分子动力学退火,退火温度为500 K~1 000 K,循环10次,获得势能面上分子构象优化,使局部能量最小。最后通过几何结构优化,获得稳态的几何构型,如图1所示。
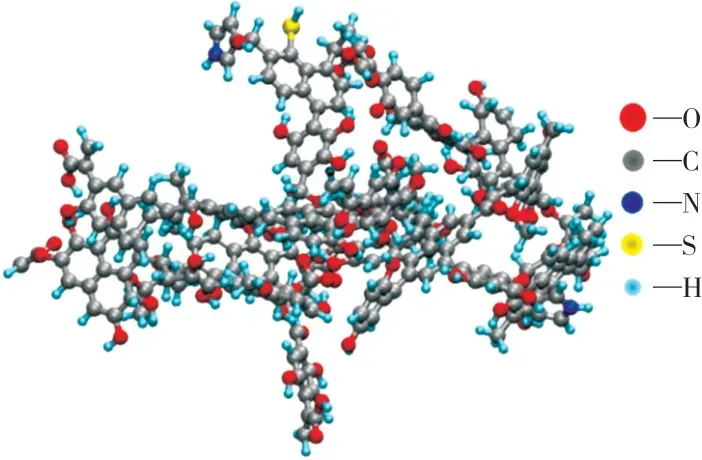
图1 优化后的宁东煤化学结构3D模型Fig.1 Optimized 3Dchemical structure model of Ningdong coal
2 热解模型构建与模拟条件设置
2.1 宁东煤热解模型构建与优化
构建宁东煤热解模型时,首先将2个优化后的宁东煤化学结构放入box构建低密度(0.5 g/c m3)结构,在LA MMPS模拟器中进行100 ps的结构弛豫优化,优化采用NPT系综,温度设置为273 K,并采用Berendsen控温器进行控温,压力为101.325 k Pa,时间步长为0.1 fs。将优化后的模型作为模拟热解过程的初始构型[20]。
经过优化,box的边长由初始的3.257 n m变为2.659 n m,内部原子排列紧密,相互聚合收缩,且没有产生新的物质(见图2)。热解模型参数优化过程曲线(见图3)显示,系统的总能量逐渐降低并稳定在-683 866 432 J/mol左右,系统密度逐渐上升后稳定在0.92 g/c m3左右。因此,可以说明获得的是稳定的煤化学结构,可用于模拟热解过程。
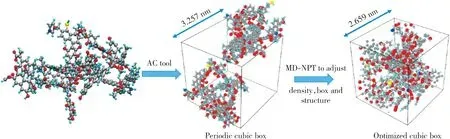
图2 宁东煤热解模型弛豫优化过程Fig.2 Relaxation optimization process of Ningdong coal pyrolysis model
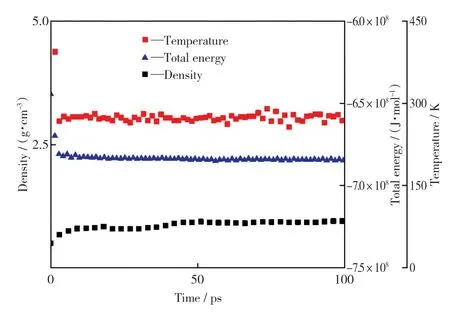
图3 热解模型参数优化过程曲线Fig.3 Opti mization process curves of pyrolysis model parameters
2.2 热解条件设置
升温速率和热解温度是影响煤热解行为和产物分布的重要因素,为考察微观路径并缩短化学反应发生周期,将上述弛豫优化后的模型在放大的模拟温度1 000 K~3 000 K范围内,分别以8 K/ps,16 K/ps,32 K/ps和64 K/ps的升温速率进行Reax FF MD模拟,分析其产物变化规律。在模拟设置上,均采用Berendsen温控器进行控温,模拟过程产物输出截断值为0.3,时间步长为0.25 fs。随后在2 500 K热解温度下进行250 ps恒温热解过程模拟。
选择8 K/ps,16 K/ps,32 K/ps和64 K/ps的升温速率主要是方便模拟步数取整,便于后续的模拟数据处理。而恒定热解温度(2 500 K)是通过分析升温过程模拟产物结果并结合文献[27]结论而确定的。截断值的选择参考了LA MMPS使用手册对Reax FF反应力场分析的建议值,取0.3通常可以获得良好且准确的结果。值得注意的是,Reax FF MD模拟温度明显高于实际实验温度,这是因为模拟过程发生在较短的时间(通常为皮秒级)内,远低于实验时间(通常为秒级)。提高模拟温度可以增加原子间的碰撞从而使得反应能够在极短的时间内发生。目前已有研究[21,30]证实温度评估策略能成功反映实验温度下的反应机理。关于总模拟时间的选取,与本研究相似的模拟工作[16,20,27]选取范围在200 ps~250 ps,因此,本研究选取250 ps作为总热解模拟时间。
3 结果与讨论
3.1 升温速率对热解产物的影响
通过统计LA MMPS输出的模拟热解产物,得到不同升温速率下热解产物总数量和种类数的变化曲线,如图4所示。
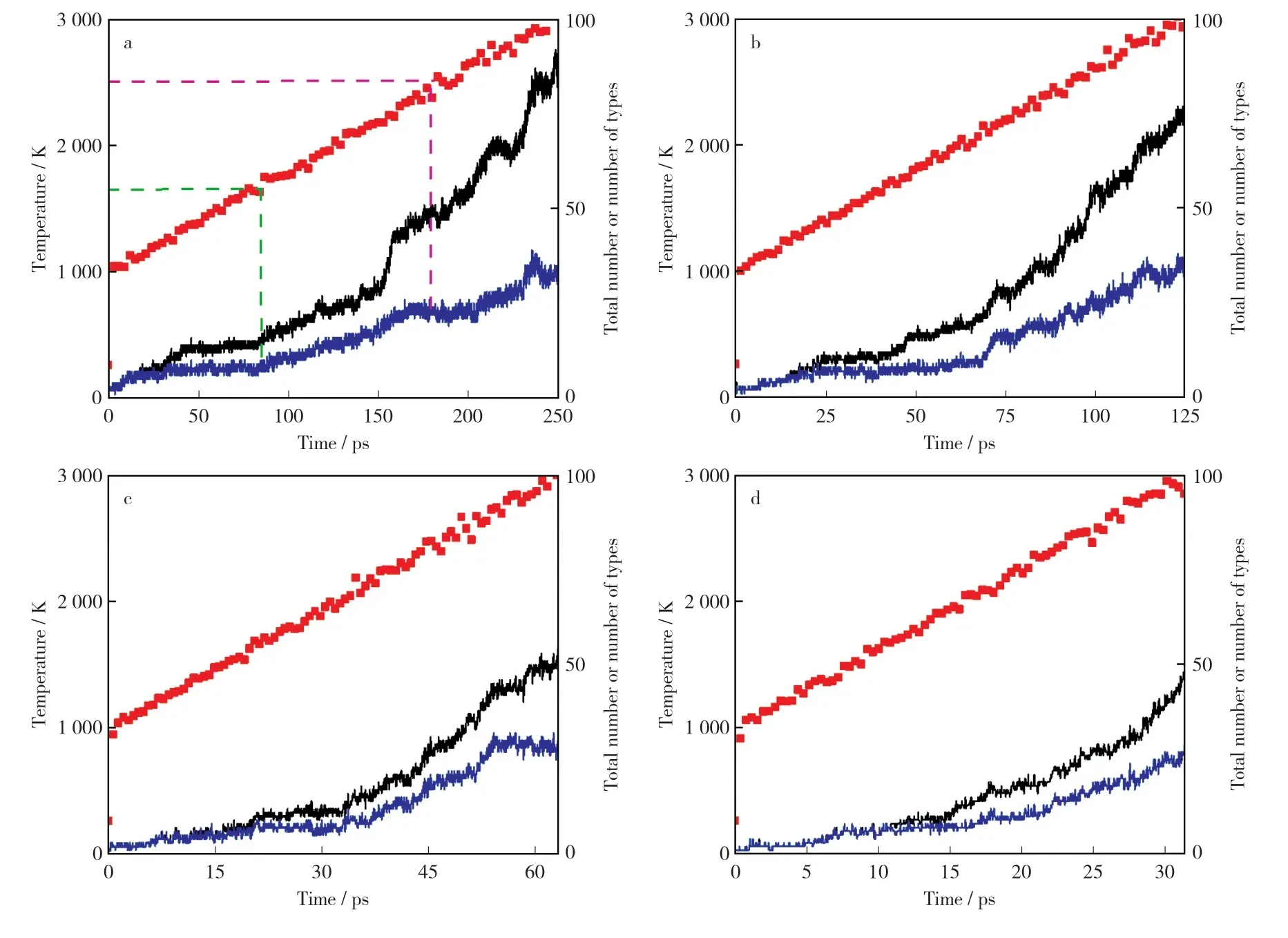
图4 1 000 K~3 000 K温度区间内不同升温速率下产物总数量和种类数的变化曲线Fig.4 Total nu mber of pr oducts and nu mber of pr oduct types under different heating rates in t he
由图4可以看出,随着温度上升,宁东煤开始发生热解,产物中出现越来越多的新碎片结构,且随温度升高总数量不断增多。大约1 500 K后热解产物种类开始明显增多,此时有大量活性自由基与有机大分子从初始结构中生成;温度升至大约2 500 K时,产物总数量和种类数快速增加,出现另一个拐点,说明模拟的热解过程开始于大约1 500 K,温度达到2 500 K之后热解反应加速,这与冯炜等[27]采用Reax FF MD方法研究枣泉煤热解所得结论一致。
对比发现4种升温速率下的最终输出产物主要包括三大部分:活性自由基(H·,OH·等)、无机气体(CO,CO2等)和多种有机物分子。为了更准确地描述热解有机产物的组成,依据C原子个数将有机物组分划分为有机气体(C1~C4)、轻质焦油产物(C5~C15)、重质焦油产物(C16~C40)和焦炭产物(C41以上)四部分。
不同升温速率和不同温度下有机物分布曲线如图5所示。由图5a可以看出,不同升温速率下热解产物主要集中于C4以下的有机气体组分。随着升温速率增大,轻质有机气体组分的数量急剧下滑,重质焦油产物数量上升,轻质焦油和焦炭产物数量整体变化不大。这说明增大升温速率会促使热解过程生成重质焦油产物。由图5b可以看出,在升温过程中,模拟温度低于1 500 K时,热解产物中气体较少,主要为重质焦油组分。随着温度的升高,气体产物快速增加,可知高温会促使大分子化合物发生二次反应,产生小分子碎片。模拟温度达到2 500 K后,重质焦油数量开始下降,有机气体和轻质焦油产量快速增加,此时生成的气、液、固三相产物比例接近实际热解过程中的相应比例。结合前文分析,2 500 K下热解反应进行较为快速,因此,2 500 K被认为是合适的热解模拟温度。
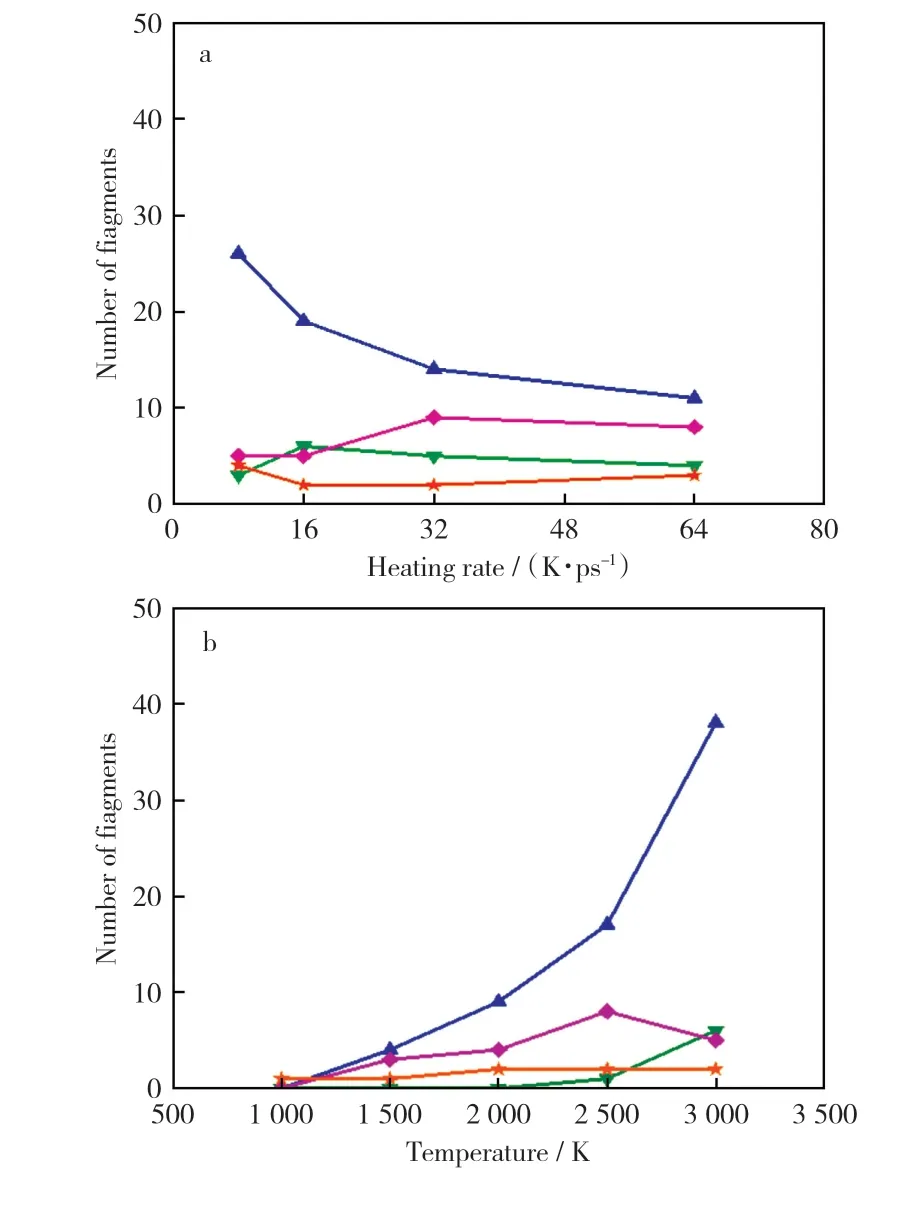
图5 不同升温速率和不同温度下有机物分布曲线Fig.5 Distribution curves of organics at different heating rates and temperatures
由于热解模型属于微观结构,且热解产物种类复杂,因此为进行粗略的定量分析,将产物中气体组分和轻质焦油产物看作高温下的具有挥发性的组分,按相对分子质量计算占比后用以对比宏观条件下工业分析中的挥发分数值,可知8 K/ps,16 K/ps,32 K/ps和64 K/ps的升温速率下,产物中具有挥发性的组分分别占28.43%,27.66%,27.93%和14.73%,而工业分析中煤干燥基的挥发分换算为干燥无灰基为27.24%,因此可以看出,16 K/ps条件下的模拟结果最接近实验值。由此可知,所建的宁东煤热解模型在合适的模拟升温速率下,模拟结果与实验数据相似,模型合理,升温速率过大会造成较大的模拟误差。
3.2 恒温热解下产物的演变
在模拟温度2 500 K下,对宁东煤模型进行250 ps的恒温热解模拟,以探究其产物分布。热解势能和热解主要产物的总数量、种类数及分布分别如图6和图7所示。
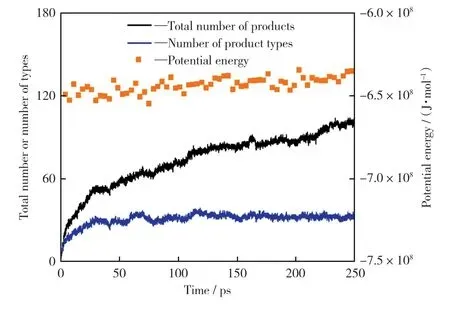
图6 2 500 K下热解势能及产物总数量和种类数Fig.6 Potential energy and total nu mber of products and nu mber of pr oduct types at 2 500 K
由图6可知,模拟过程中前50 ps产物的种类数和总数量快速上升,该阶段产生的大量气体组分多是无机气体组分(H2O,CO,CO2等),其次是有机气体组分(见图7)。主要原因是热解初期键能较弱如C—O键和桥键等优先断裂,释放出有机小分子和大量活性自由基(OH·,H·等),活性自由基间可快速反应生成气体产物。50 ps之后,热解产物种类基本平稳,但热解产物总数量不断增多,主要是无机气体增多,结合图6中系统势能的上升,说明热解体系的反应仍未结束,还存在吸收热量的二次反应,即生成的活性自由基和热解产物之间均可以两两反应,也进一步促进热解过程的进行,这与BHOI et al[21]对澳大利亚Mor well煤热解的Reax FF MD模拟结论一致。
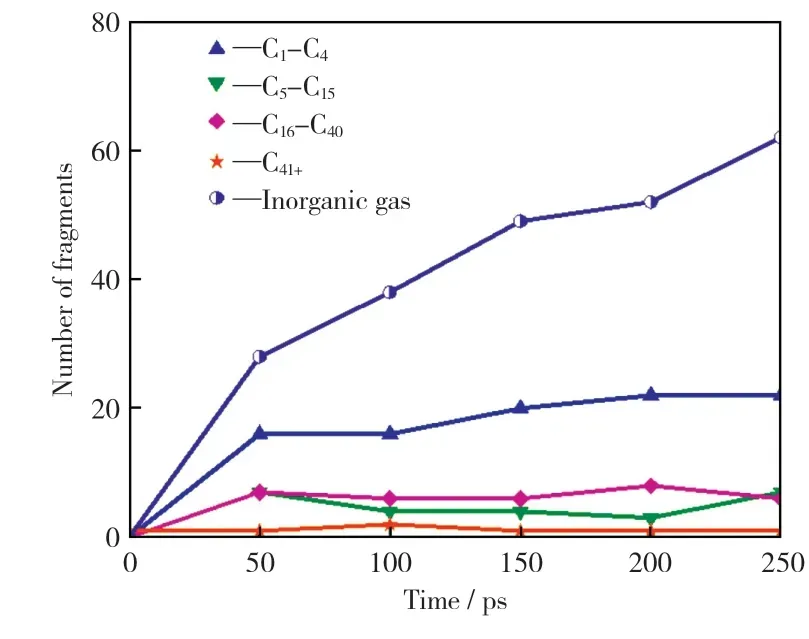
图7 2 500 K下热解产物分布曲线Fig.7 Distribution cur ves of pyrolysis products at 2 500 K
图6 和图7从能量角度和产物数量角度表明宁东煤热解模型在高温热解条件下除热解过程外还会发生二次反应,这与已知的实际煤热解过程相似,再次证明了该热解模型具有合理性。
3.3 硫元素迁移机理
为探究升温速率对S原子迁移的影响,得到宁东煤化学结构热解后的含S产物,结果见表5。该宁东煤化学结构在2 500 K,250 ps恒温热解模拟下的最终输出产物见表6。
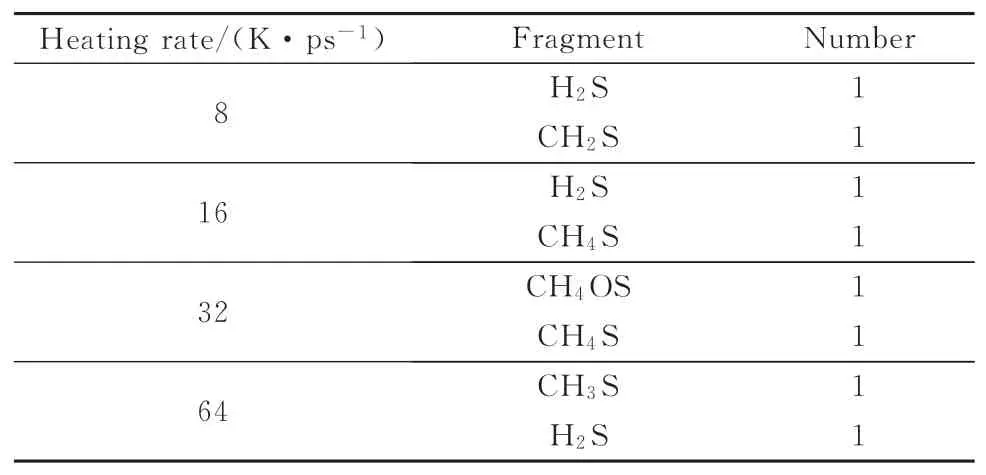
表5 不同升温速率下最终含硫产物Table 5 Final sulf ur-containing products at different heating rates
由表5可知,不同升温速率下的S原子最终均迁移到了有机小分子或者H2S中,以气态的形式存在,而表6显示,恒温热解下S原子最终都转移到了H2S气体中,这与赵丽红等[31-32]的实验结论相似。通过综合分析热解含硫产物的演变过程,得到了S原子从煤中迁移到H2S中的路径(如图8所示)。
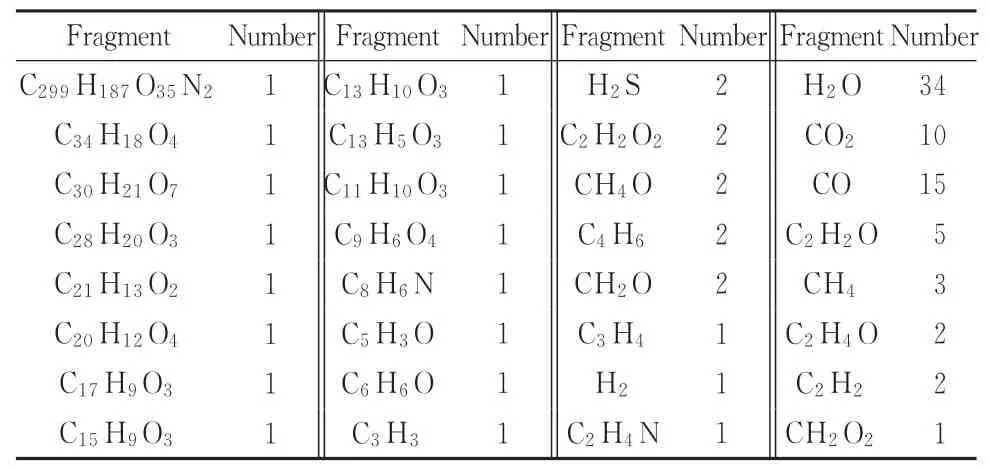
表6 2 500 K时宁东煤的热解产物Table 6 Pyrolysis products at 2 500 K
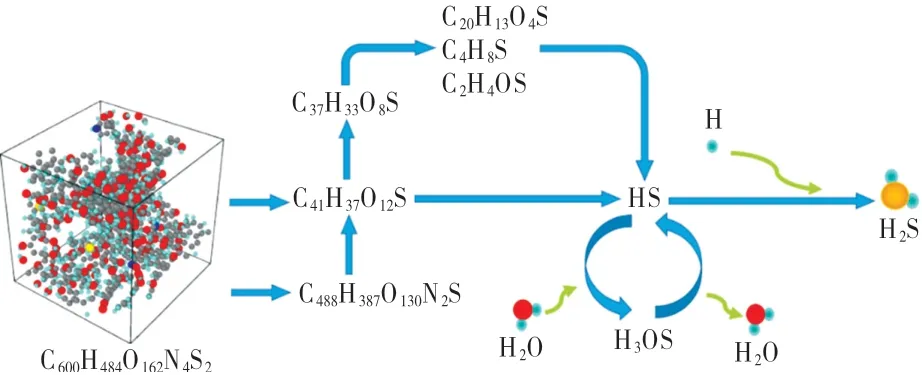
图8 宁东煤模型热解过程S原子迁移路径Fig.8 Migration pat h of sulf ur in t he pyrolysis process of Ningdong coal model
在热解模拟开始的初期(1 ps~5 ps),S原子会因系统中不稳定键的断裂而迁移到相对分子质量较小的焦炭碎片中;随着进一步热解,焦炭碎片裂解,此时S原子迁移到重质焦油中,或者从焦炭碎片中解离出来以硫氢根(HS)的形式存在;但是硫氢根不稳定,其与热解产生的H2O形成不稳定的H3OS后又在极短时间内变回硫氢根;而当硫氢根接触到H自由基时,便会生成H2S而稳定存在,直至热解过程结束;如果在焦炭碎片裂解过程S原子迁移到重质焦油中,则会随重油裂解而进入轻质焦油或者有机气体中去。同时可以发现S原子的活跃性,重质焦油裂解期间,出现了S原子迁移到相对分子质量更大的碎片中,但在很短时间内又迁移出来的现象。在总体模拟时间轴上S原子逐渐会往相对分子质量更小的碎片中转移,最终以硫氢根的形式解离出来,直至与H自由基结合形成H2S。H2S化学性质非常活跃,遇到氧极易反应生成SO2(和水),从而成为烟气中的有害气体组分。
4 结 论
1)升温速率过高,会促进重质焦油的产生,不利于有机气体裂解,导致热解产物种类数和总数量下降,热解结果偏离实际值较大。
2)热解反应主要从模拟温度1 500 K开始,2 500 K下热解剧烈,2 500 K是合适的宁东煤热解的模拟温度。
3)在2 500 K模拟条件下宁东煤热解模型热解状况合理并发生二次反应,与实际实验的煤热解过程特点相似,证明了该模型的合理性,模拟结果的机理分析具有参考价值。
4)升温速率对宁东煤中S元素迁移影响不大,S元素均会转移至H2S或者含硫有机气体中。宁东煤中巯基形式存在的S原子在热解过程中容易迁移到相对分子质量小的碎片中,最终将以硫氢根的形式与H自由基结合生成H2S,进而参与后续化学链燃烧等反应。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
恋爱婚姻家庭·青春(2019年11期)2019-12-11
恋爱婚姻家庭(2019年32期)2019-11-19
小雪花·初中高分作文(2017年10期)2018-05-15
今日财富(2017年32期)2017-10-19
中学生数理化·高二版(2008年5期)2008-11-12
