
柱前衍生法测定岩藻多糖中的岩藻糖含量
2022-02-06 07:00周宏霞
农产品加工 2022年23期
周宏霞,王 楠,都 颖,王 妙,张 羽
(威海市食品药品检验检测研究院,山东 威海 264200)
岩藻多糖也称褐藻多糖硫酸酯,是以岩藻糖和硫酸基为主要特征的一类水溶性多糖。Kylin在1913年首次从掌状海带中提取,并命名为Fucoidin,现在已统一定名为岩藻多糖(Fucoidan)或岩藻聚糖硫酸酯(Sulfated Fucan)[1]。以海带、裙带菜等褐藻为原料,经精制提取可以得到多糖类产品——岩藻多糖[2]。岩藻多糖具有多种生物活性,包括抗肿瘤、抗凝血、抗病毒、降血糖、降血脂、免疫调节等活性,是一种重要的生物活性物质[3-5]。L-岩藻糖是影响岩藻聚糖生物活性的重要组成成分,岩藻聚糖硫酸酯的重要指标性成分。而且L-岩藻糖作为岩藻聚糖的特征单糖,常用于衡量岩藻聚糖的纯度[6]。因此,在测定褐藻制品中岩藻糖含量时,以测定L-岩藻糖的含量来表示。目前,检测岩藻糖的方法有气相色谱法[7-9]、高效液相色谱-柱前衍生化紫外检测法[10]或蒸发光法[11]、离子色谱法[6]等。其中,高效液色谱-柱前衍生化紫外检测法用的比较广泛。在SC/T 3404—2012中有关于岩藻糖测定的方法,但是应用该方法进行衍生,生成的衍生物质不大稳定,造成结果重现性不好。在该方法的基础上进行了改进与研究,建立了一种比较稳定的柱前衍生-高效液相色谱法测定L-岩藻糖的方法。
1 仪器与试剂
1.1 试验试剂
乙腈、甲醇(色谱纯,Thermo);磷酸二氢钾(优级纯);1-苯基-3-甲基-5-吡唑啉酮(PMP)(≥99.0%,麦克林);三氟乙酸(优级纯);氢氧化钠(优级纯);三氯甲烷(色谱纯);冰乙酸(色谱纯);盐酸(优级纯);氨水(分析纯);L-岩藻糖(L/Stanford Chemicals,≥99.5%);试验用水为去离子水。
1.2 试剂配制
4 mol/L三氟乙酸溶液:准确量取三氟乙酸溶液29.7 mL,加水定容至100 mL。4 mol/L氢氧化钠溶液:准确称取氢氧化钠16.0 g,加水溶解并定容至100 mL。0.1 mol/L磷酸二氢钾缓冲溶液:准确称取磷酸二氢钾1.36 g,加水80 mL溶解,用氨水调节pH值为6.0,再用水定容至100 mL。0.5 mol/LPMP甲醇溶液:准确称取PMP 8.71 g,用甲醇溶解并定容至100 mL。0.3 mol/L氢氧化钠溶液:准确称取1.2 g氢氧化钠,加水溶解并定容至100 mL。0.3 mol/L盐酸溶液:准确移取盐酸溶液2.5 mL,用水稀释至100 mL。0.05 mol/L磷酸二氢钾缓冲液:准确称取磷酸二氢钾6.8 g,加80 mL水溶解,用氢氧化钠溶液调节pH值为6.0,再用水定容至1 000 mL,备用。L-岩藻糖溶液配制:精密称取L-岩藻糖10 mg,用水溶解定容至10 mL,配制成浓度为1.0 mg/mL的储备液。
1.3 仪器
高效液相色谱仪,配二极管阵列检测器;分析天平;涡旋混合器;电热恒温干燥箱;恒温水浴锅;超声波清洗器,Mili-Q超纯水机。
2 试验方法
2.1 色谱条件
色谱柱(C18柱,Venusil MP 4.6 mm×250 mm,5 μm,艾杰尔科技有限公司),柱温为40℃,流速为1.0 mL/min,流动相为0.05 mol/L磷酸盐缓冲液(pH值6.8)-乙腈(体积比为85∶15),检测器:二极管阵列检测器,波长254 nm,进样量为20 μL。
2.2 岩藻多糖的水解
准确称取岩藻多糖样品0.1 g(精确至0.000 1 g)于水解管中,加入4 mol/L三氟乙酸溶液10 mL,混匀后充入氮气封盖,在110℃电热恒温干燥箱中水解2 h,取出后冷却至室温,加入4 mol/L氢氧化钠溶液10 mL中和,调节pH值至中性。将水解液转移到25 mL容量瓶中,用0.1 mol/L磷酸二氢钾缓冲溶液定容至刻度,备用。根据样品中岩藻糖含量的不同可以进行不同浓度的稀释。
2.3 试样的衍生及净化
准确移取定容后的水解液450 μL于10 mL塑料离心管中,加入0.5 mol/L的1-苯基-3-甲基-5-吡唑啉酮(PMP)甲醇溶液450 μL和0.3 mol/L氢氧化钠溶液450 μL,涡旋混合,70℃水浴反应70 min,取出冷却至室温,加0.3 mol/L盐酸溶液450 μL,涡旋混合,加三氯甲烷溶液2 mL,充分振荡,静置分层,吸弃下层三氯甲烷层,重复萃取2次。上清液过0.45 μm的滤膜,供高效液相色谱仪分析。
2.4 标准溶液的衍生化
准确移取适量的标准储备溶液,用水稀释成1,5,10,50,100,200,500 μg/mL的标准工作液,分别取450 μL于10 mL塑料离心管中,按照2.3的操作进行衍生化,上机检测,绘制校准曲线。
3 结果与分析
3.1 衍生条件的优化
采用相同的衍生液、衍生温度和衍生时间进行试验发现,用盐酸溶液中和的稳定性要优于醋酸溶液中和。王泽文等人[10]对岩藻糖的衍生温度和衍生时间进行研究发现,采用70℃反应70 min能充分反应完全。因此采用了70℃水浴反应70 min。衍生反应后,由于PMP试剂在波长245 nm处有很大的紫外吸收,为防止试验中PMP残留对衍生物检测的干扰,在试验中用三氯甲烷去除剩余的PMP。
3.2 色谱条件的优化
采用磷酸盐缓冲液(pH值6.8)-乙腈(体积比为85∶15)为流动相,在该条件下目标物质和杂质能很好地分离。检测器采用二极管阵列检测器,采集光谱图,通过顶点的光谱图的不同,可以很好地区分岩藻糖和杂质。
岩藻糖衍生物谱图见图1,岩藻糖衍生物光谱图见图2。

图1 岩藻糖衍生物谱图
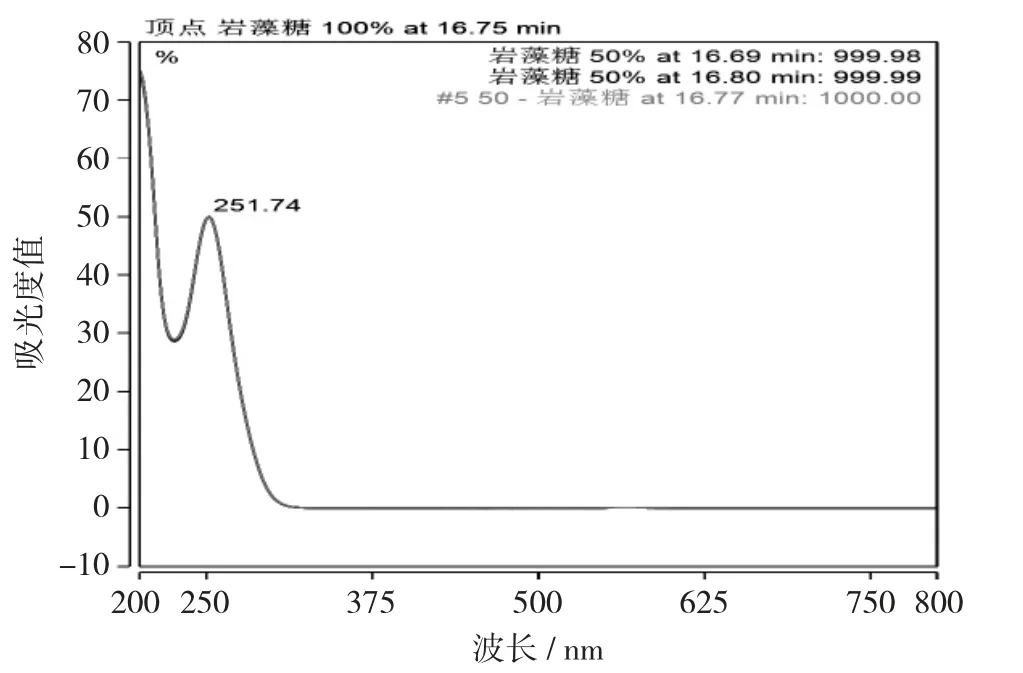
图2 岩藻糖衍生物光谱图
3.3 线性关系考查
按照2.1色谱条件,分别测定L-岩藻糖系列标准工作溶液,每个浓度测定1次,记录L-岩藻糖色谱峰面积,以L-岩藻糖系列标准工作溶液的质量浓度X(μg/mL)为自变量、色谱峰面积Y为因变量,进行线性回归,得线性方程:Y=0.673 9X+2.281 6,相关系数R2为0.999,表明L-岩藻糖的质量浓度在1~500 μg/mL范围与色谱峰面积线性关系。
岩藻糖校准曲线见图3。
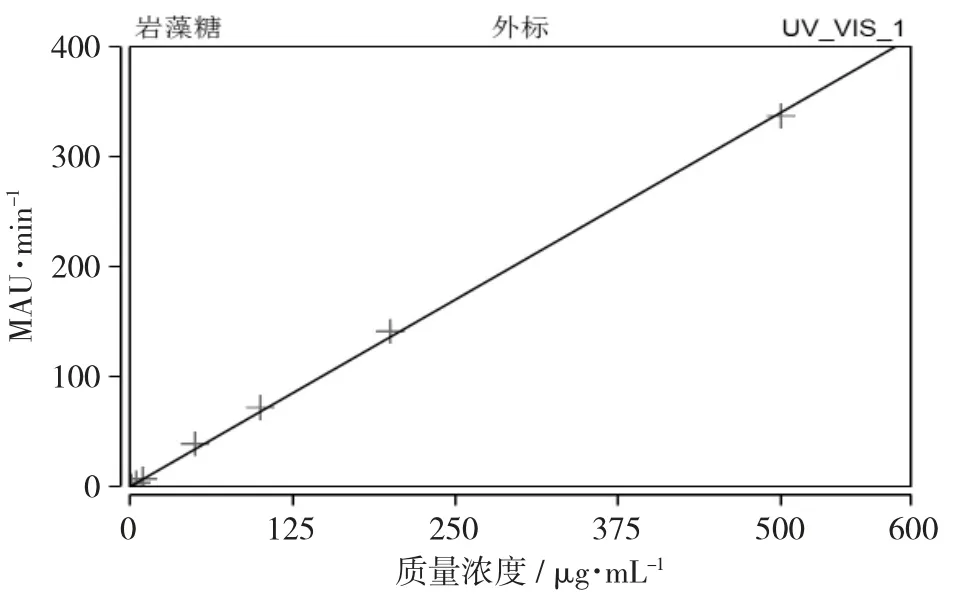
图3 岩藻糖校准曲线
3.4 精密度试验(n=6)
取100 μg/mL L-岩藻糖,按照2.3方法进行衍生,精密吸取20 μL该溶液,进样后测得色谱峰面积,连续重复进样6次,计算其相对标准偏差。结果为:RSD为0.73%,说明仪器有良好的精密度。
3.5 稳定性试验结果(n=6)
称取100 μg/mL L-岩藻糖,按照2.3方法进行衍生,精密吸取20 μL该溶液,在0,4,8,10,12,24 h依次进样一次,测定峰面积值,连续进样6次,计算其相对标准偏差,结果为:RSD为0.58%。说明该对照品溶液的衍生化在24 h有良好的稳定性。
3.6 重复性试验结果(n=6)
准确称取岩藻多糖样品0.1 g 6份样品,按照本文的方法进行水解和衍生,计算相对标准偏差。结果为:2.68%,说明该方法具有良好的重复性。
3.7 加标回收率试验
准确称取岩藻糖浓度为15.6%的岩藻多糖样品0.1 g 6份,加入岩藻多糖标准溶液,使样品中的加标浓度为15%,按照试验方法进行水解和衍生,计算加标回收率和相对标准偏差,分别为94.7%和5.27%。
岩藻糖回收率试验见表1。
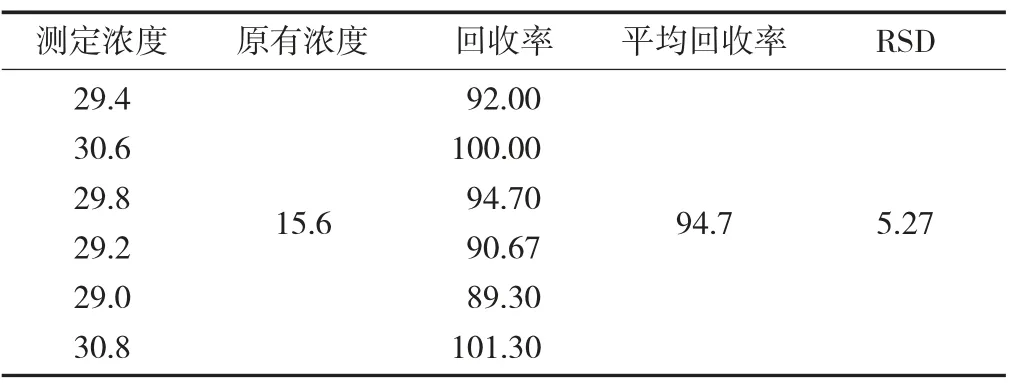
表1 岩藻糖回收率试验/%
4 结论
采用PMP柱前衍生法和高效液相色谱法对岩藻多糖中的L-岩藻糖质量浓度进行了测定分析,结果表明,该方法测定L-岩藻糖质量浓度简便、快捷,结果的稳定性,重复性好,测定结果准确。PMP衍生单糖后使其带上发色基团,从而有紫外吸收,采用二极管阵列检测器能得到样品中物质的光谱图,可以很好地区分岩藻糖衍生物和杂质。通过该方法的测定,可以得到岩藻多糖中L-岩藻糖的含量,从而判定该产品是否符合岩藻多糖的产品标准,达到对岩藻多糖产品质量的控制。
猜你喜欢
食品与发酵工业(2022年7期)2022-04-18
农业科技通讯(2021年4期)2021-05-23
食品与生物技术学报(2021年11期)2021-01-17
天然产物研究与开发(2020年10期)2020-11-10
食品研究与开发(2020年16期)2020-08-11
中国化肥信息(2019年5期)2019-06-25
天然产物研究与开发(2019年4期)2019-04-27
山东工业技术(2019年6期)2019-03-27
科技创新导报(2017年20期)2017-09-13
中国化肥信息(2017年4期)2017-06-24
