
Nd改性CeO2/GO催化剂对NH3-SCR抗硫性能的影响
2022-02-04 08:02袁群富周志元郭晏乐辛志玲李瑾
应用化工 2022年12期
袁群富,周志元,郭晏乐,辛志玲,李瑾
(上海电力大学 上海市电力材料保护与先进材料重点实验室,上海 200090)
氮氧化物(NO和NO2)会导致酸雨和光化学烟雾,危害人体健康。目前,NH3选择性催化还原(NH3-SCR)技术是最高效的NOx脱除方法[1],世界范围内已有大量研究报道了低温NH3-SCR催化剂,但提高催化剂抗SO2中毒能力仍是目前研究的重点[2-4]。稀土金属(Nd、Nb、Dy、Sm、Eu、Sb等)引入到NH3-SCR催化剂中,是提高抗SO2中毒能力的一种实用方法[5-11]。
本文通过一步浸渍法制备了Nd改性的CeO2/GO催化剂。Nd0.5CeOx/GO在300 ℃和100 mg/L SO2条件下具有较强的抗SO2性能。TEM、SEM、XRD、XPS、BET和FTIR的表征结果表明,高度分散的Nd在CeO2/GO催化剂上可以减少活性相Ce的硫酸化。
1 实验部分
1.1 试剂与仪器
氧化石墨烯(GO)、硝酸铈、硝酸钕均为分析纯;实验用水为超纯水(Milli-Q,18.2 MΩ·cm);一氧化氮(3%NO,97%N2)、氨气(3%NH3,97%N2)、氮气、氧气(99.999%)、二氧化硫(3%SO2,97%N2)、氦气(99.999%)等气体。
DF-101S 型集热式恒温加热磁力搅拌器;SCQ-250A 型超声波清洗仪器;SK-GO8123K 型气氛管式电炉;KMN-SCR-32型微型反应装置;SMC-9021型烟气排放连续监测系统;FEI Tecnai G2 F20型透射电子显微镜;JSM7610F型扫描电子显微镜;SmartLab 型X射线衍射仪;ESCALAB 250Xi 型X射线光电子能谱仪;ASAP2460 3.01 型全自动比表面及孔隙度分析仪;Spectrum Two 型傅里叶红外漫反射分析仪。
1.2 催化剂的制备
将900 mg氧化石墨烯溶解在120 mL超纯水中,超声15 min,得到均匀的黑色溶液。将129 mg的Ce(NO3)2·6H2O和一定量的Nd(NO3)3·6H2O分散在15 mL超纯水中,超声处理5 min,得到前驱体溶液。将前驱体溶液滴加到GO溶液中,超声30 min 。将黑色混合物溶液置于90 ℃水浴中搅拌,直至干燥。用管式炉在He氛围中以10 ℃/min的升温速率在500 ℃下煅烧4 h,得到Nd改性CeO2/GO催化剂。根据摩尔比Nd2O3/(Nd2O3+CeO2) =0.3,0.4,0.5,0.6,分别命名为Nd0.3CeOx/GO、Nd0.4CeOx/GO、Nd0.5CeOx/GO和Nd0.6CeOx/GO。
为了比较,通过相同的工艺制备了CeO2/GO催化剂。
1.3 催化剂表征
通过透射电子显微镜和扫描电子显微镜确定催化剂的形貌结构。用X射线衍射仪对样品的晶体结构进行分析。通过X射线光电子能谱仪测定催化剂表面元素价态及含量。利用全自动比表面及孔隙度分析仪分析样品的物理性质。通过傅里叶红外漫反射分析仪测定新鲜和SO2中毒催化剂上形成的表面物质。
1.4 催化剂性能评价
选择内径为10 mm的固定床SCR系统,进行催化剂抗硫性能测试。反应气体的组成如下:500 mg/L NO,550 mg/L NH3,100 mg/L SO2、3% O2,N2作为平衡气,总流速为600 mL/min,空速(GHSV)为15 000 h-1。NO转化率计算公式为:

公式中的NOin和NOout分别是反应器入口和出口的NO浓度。
2 结果与讨论
2.1 催化剂SCR抗硫性评价
Nd改性CeO2/GO催化剂在300 ℃下进行了6 h 的抗SO2中毒测试,结果见图1。
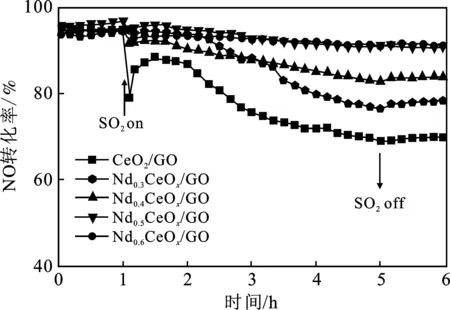
图1 所有催化剂的抗SO2中毒性能Fig.1 Resistance to SO2 poisoning of all catalyst samples
由图1可知,所有样品的NO转化率在添加100 mg/L SO2后均有所下降。在1 h的连续反应中,CeO2/GO的NO转化率从90%降到77.84%,Nd0.5CeOx/GO从91.4%降到91.0%,4 h后CeO2/GO脱硝性能降到48.08%,Nd0.5CeOx/GO的NO转化率迅速稳定在84.9%。关闭100 mg/L SO2后,NO转化率略有增加,这可能与催化剂表面硫酸铵的分解有关。
2.2 扫描电子显微镜(SEM)及透射电子显微镜(TEM)
通过透射电子显微镜和扫描电子显微镜对Nd0.5CeOx/GO催化剂的形貌、组成和结构进行分析,通过EDX分析了催化剂样品中C、O和Nd元素的分布,结果见图2。

图2 Nd0.5CeOx/GO催化剂的微观形貌Fig.2 The microscopic morphology of Nd0.5CeOx/GO catalyst(a) Nd0.5CeOx/GO催化剂的HAADF-STEM图像;(b)~(e) C、O、Ce和Nd的相应EDX元素图;(f) Nd0.5CeOx/GO的TEM图像;(g) Nd0.5CeOx/GO的扫描电镜图像
由图2(a)和图2(f)可知,在负载Nd和Ce元素后Nd0.5CeOx/GO催化剂仍保持着薄片状的形态,证明了其二维片状特征。由图2(b~e)可知,C、O、Ce和Nd元素分布均匀,表明Nd和Ce均匀地负载在GO表面。由催化剂的SEM图像图2(g)可知,大量的Ce和Nd纳米颗粒分散堆叠在波纹状的GO上。
2.3 X射线衍射(XRD)分析
通过X射线衍射(XRD)对所有催化剂样品的晶体结构进行分析,结果见图3。
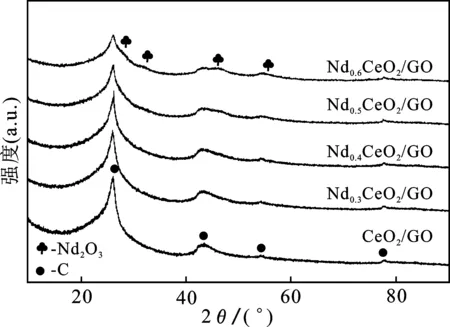
图3 所有催化剂的 XRD 图谱Fig.3 XRD patterns of all catalysts
由图3可知,CeO2/GO催化剂的衍射峰位置分别为26.38,42.22,54.54,77.24 °,分别对应于石墨烯的(002)、(100)、(004)和(110)晶面(PDF #41-1487)。一般认为,XRD衍射峰越尖锐代表催化剂的结晶度越高,说明催化剂表面晶格缺陷越少,并且这将使催化剂表面的活性位点难以暴露[12]。随着Nd的加入,26.38°的衍射峰强度逐渐降低,逐渐可以观察到Nd2O3的衍射峰,说明Nd元素的加入降低了GO的结晶度,有利于催化剂暴露更多的活性位点。Nd2O3的衍射峰27.85,32.26,46.30,54.93 °分别对应于Nd2O3的(222)、(400)、(440)和(622)晶面(PDF #21-0579)。所有样品均未检测到CeO2的衍射峰,这可能是因为CeO2高度分散在催化剂表面或以无定形状态存在。
2.4 X射线光电子能谱(XPS)
图4为Nd改性CeO2/GO催化剂的XPS分析结果,表1列出了表面元素的浓度。
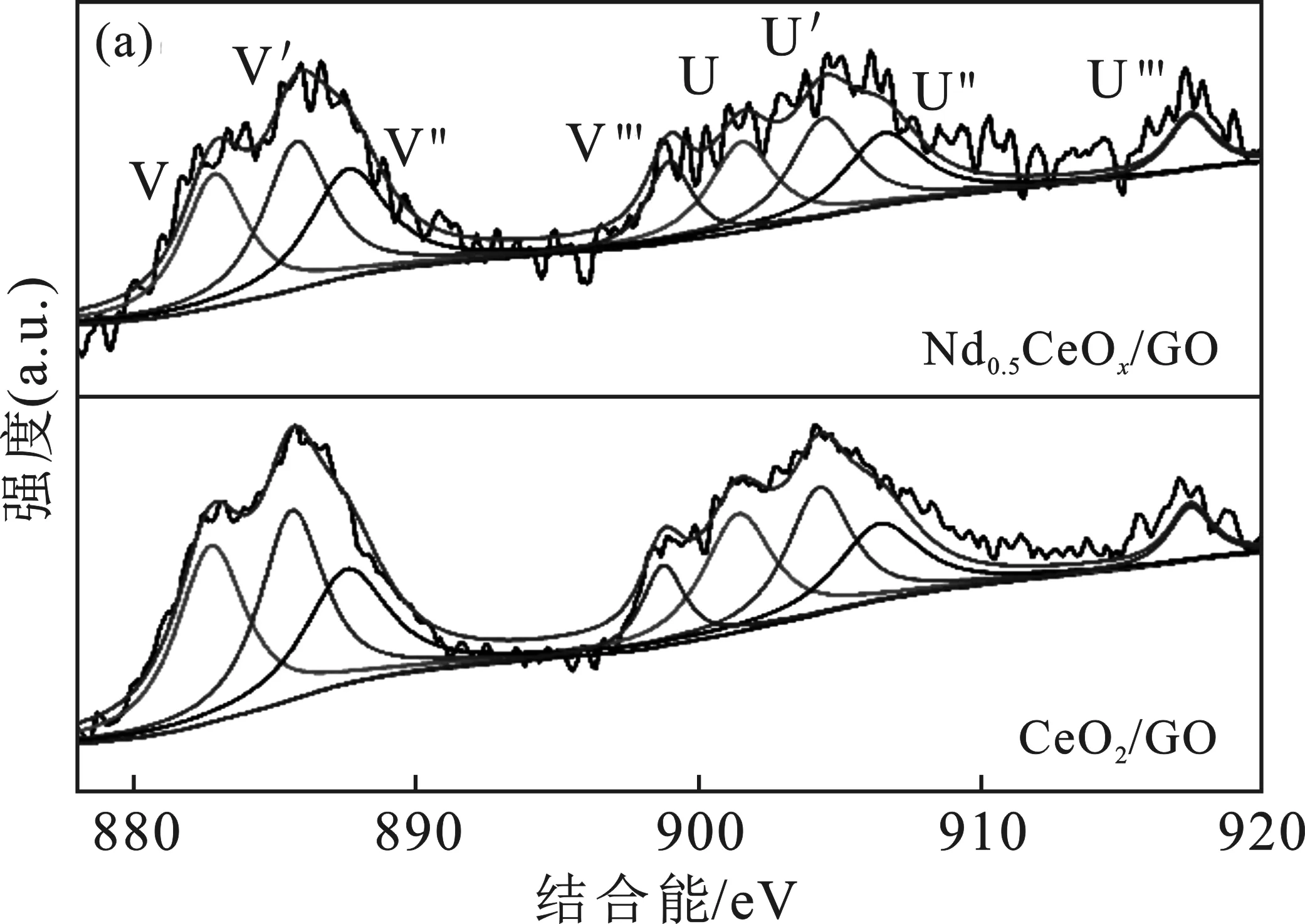
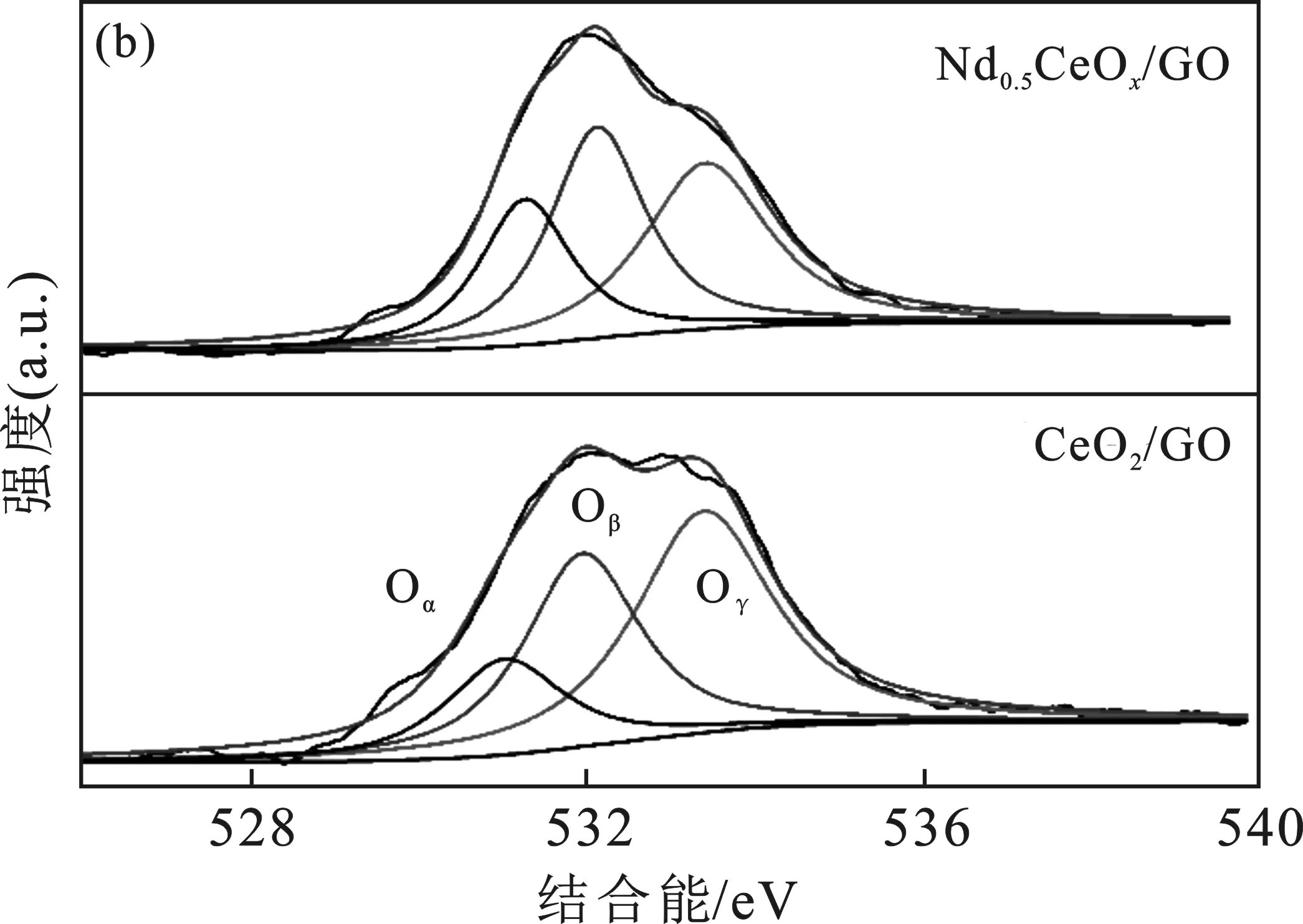
图4 (a) CeO2/GO和Nd0.5CeOx/GO的Ce 3 d 轨道的XPS光谱,(b) CeO2/GO和Nd0.5CeOx/GO 的O 1 s轨道的XPS光谱Fig.4 (a) XPS spectra of Ce 3 d orbitals of CeO2/GO and Nd0.5CeOx/GO,(b) XPS spectra of O 1 s orbitals of CeO2/GO and Nd0.5CeOx/GO
由图4(a)可知,Ce 3 d3/2和Ce 3 d5/2的峰分别标记为U和V。在885.6 eV和904.2 eV处标记为V’和U’的特征峰表示3d104f1的初始电子状态,对应于Ce3+[13]。Ce4+的3d104f0电子态分别标记为U、U″、U‴、V、V″和V‴[14]。由表1可知,CeO2/GO和Nd0.5CeOx/GO催化剂的Ce3+比例分别为33.3%和32.0%。Ce3+的存在会引起催化剂表面电荷不平衡,产生氧空位,形成化学吸附氧,从而提高催化性能[15]。因此,较高的Ce3+/(Ce3++Ce4+)相对原子浓度比可以促进NO向NO2的转化并促进“快速”SCR反应(NO+NO2+2NH3→2N2+3H2O)[15]。
由图4(b)可知,O 1 s峰可以拟合为3个特征峰,其中晶格氧(Oα)归因于531.2 eV,化学吸附氧(Oβ)归因于532.3 eV,羟基氧(Oγ)归因于533.7 eV[16]。与Oα相比,催化剂表面的Oβ具有更高的电子转移率,有利于加速NO氧化为NO2[17]。因此,人们普遍认为吸附氧在氧化反应体系中具有更强的反应性。见表1,Nd改性后催化剂上的Oβ比值[定义为Oβ/(Oα+Oβ+Oγ)]从30.2%增加至37.0%。因此,Nd的添加有利于提高CeO2/GO催化剂的Oβ比。
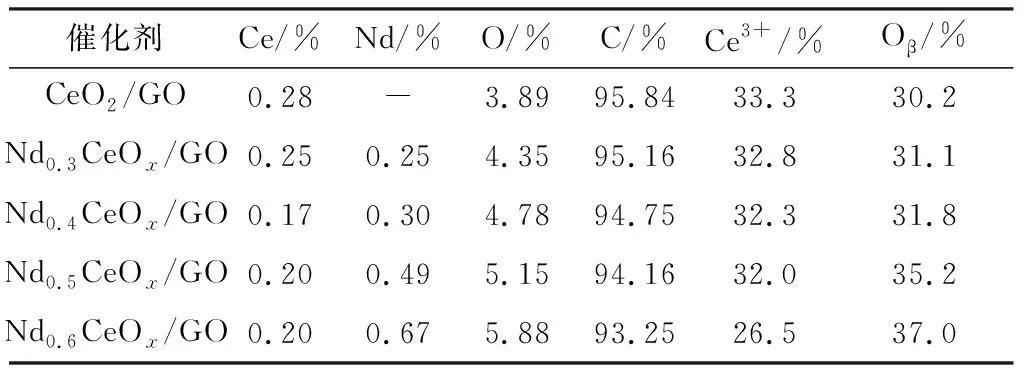
表1 催化剂表面元素含量Table 1 Surface element contents of catalysts
2.5 BET分析
图5(a)为Nd0.5CeO2/GO催化剂的N2吸附脱附曲线和孔径分布曲线,图5(b)显示了GO、CeO2/GO和Nd0.5CeOx/GO的N2吸附脱附曲线。
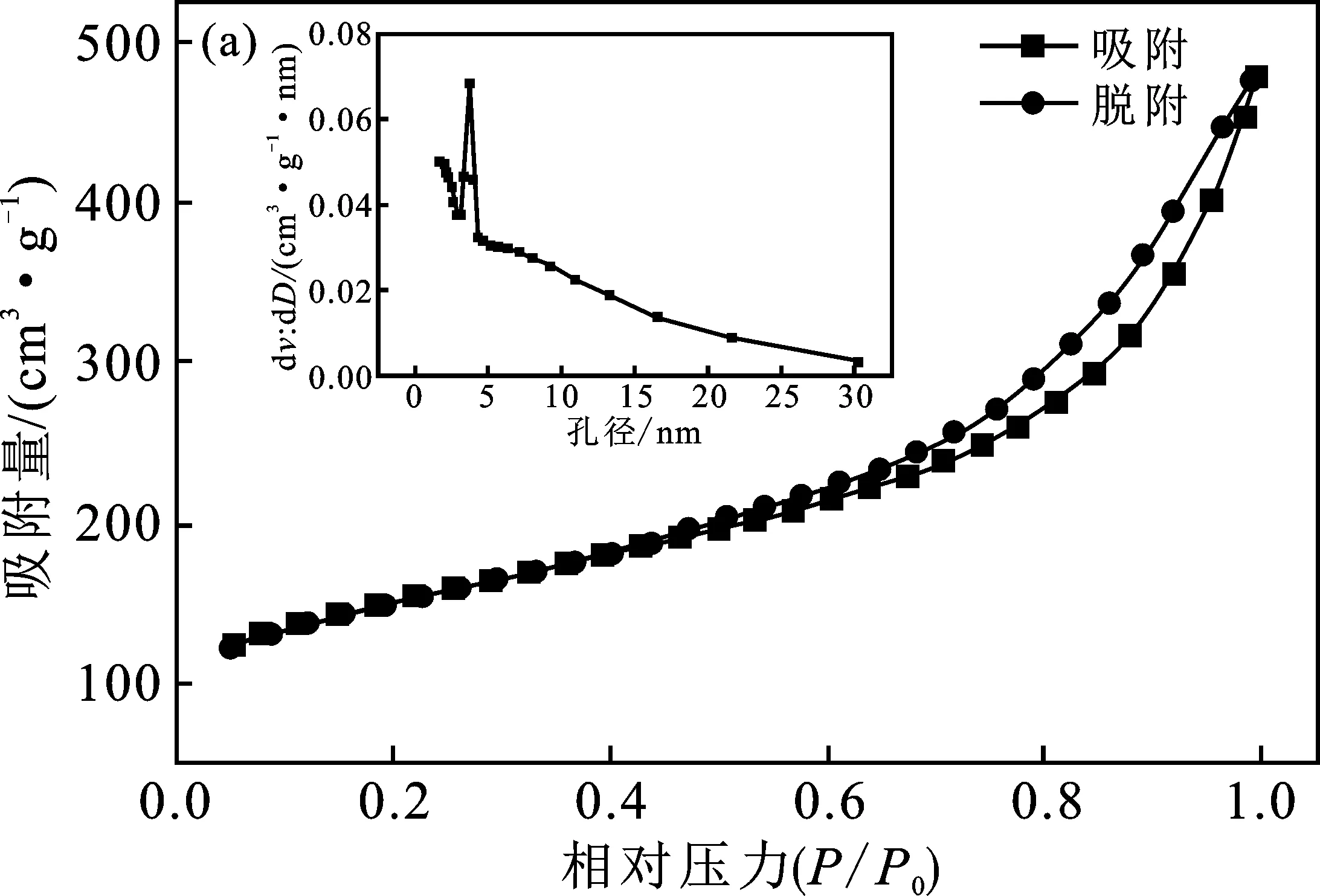
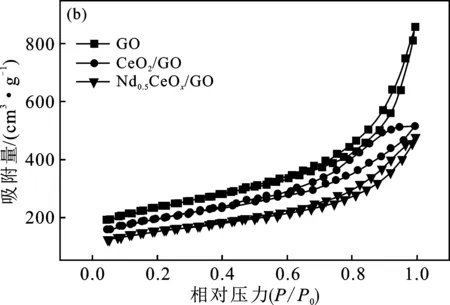
图5 (a)Nd0.5CeOx/GO的BET分析;(b) GO,CeO2/GO 和Nd0.5CeOx/GO的N2吸脱附曲线Fig.5 (a) BET analysis of Nd0.5CeOx/GO;(b) N2 adsorption and desorption curves of GO,CeO2/GO and Nd0.5CeOx/GO
由图5可知,根据IUPAC的分类,在0.45~1.0的相对压力P/P0范围内吸附脱附曲线呈现典型的IV型等温线和H3滞后回线[18],这表明催化剂具有更多的介孔结构。根据其孔径分布,其孔径主要为2~30 nm,属于介孔[19]。由表2可知,Nd0.5CeO2/GO催化剂的比表面积为528.83 m2/g,平均孔径为5.58 nm。随着GO上负载的活性相增加,催化剂样品的比表面积和孔体积减少,这可能是因为活性组分堵塞了GO的孔隙[20]。此外,与CeO2/GO相比,Nd0.5CeO2/GO的平均孔径有所增大,这可能是因为掺杂Nd有助于提高活性相(Nd和CeO2)在载体表面分散度,从而降低了孔隙的堵塞程度[21]。Nd0.5CeO2/GO催化剂的比表面积虽然下降到528.83 m2/g, 但仍具有较高的比表面积,有利于促进反应气体的吸附,提高SCR的催化活性。
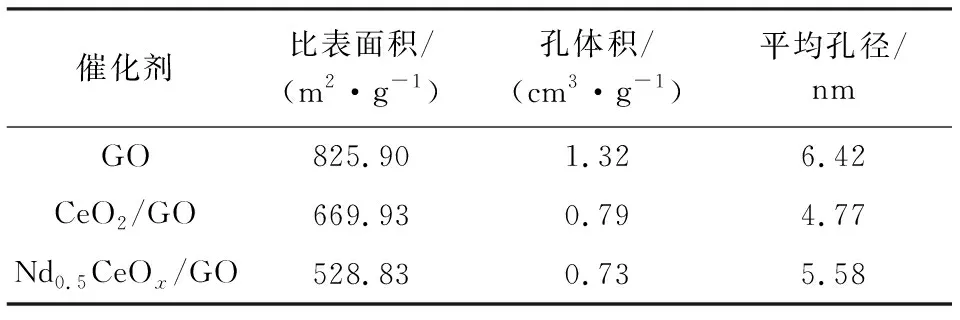
表2 GO、CeO2/GO和 Nd0.5CeOx/GO的结构性质Table 2 Structural properties of GO,CeO2/GO and Nd0.5CeOx/GO
2.6 傅里叶红外光谱(FTIR)分析
用FTIR研究了催化剂表面官能团在抗SO2中毒测试后的变化,结果见图6。
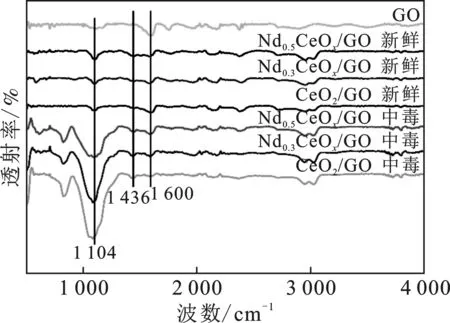
图6 GO载体、新鲜和SO2中毒的CeO2/GO催化剂、新鲜 和SO2中毒的Nd0.3CeOx/GO催化剂和新鲜 SO2中毒的Nd0.5CeOx/GO催化剂的FTIR图Fig.6 FTIR diagram of GO carrier,fresh and SO2 poisoned CeO2/GO catalyst,fresh and SO2 poisoned Nd0.3CeOx/GO catalyst and fresh and SO2 poisoned Nd0.5CeOx/GO catalyst

2.7 离子极化理论
根据离子极化理论[25],金属硫酸盐的形成可视为其分解的逆过程。
MeSO4(S)←→MeO(S)+SO3
其中,Me代表金属。
即CeOx/GO催化剂掺杂了另一种金属元素Nd,其硫酸盐的分解温度高于硫酸铈的分解温度,则SO2将优先与金属元素Nd结合,形成相应的硫酸盐,从而保护CeOx不被硫酸化。事实上,文献中记载的Nd2(SO4)3的分解温度大于1 000 ℃,高于Ce2(SO4)3的分解温度973 ℃[11,26]。因此,与CeOx相比,催化剂表面的NdOx更容易被硫酸化,从而抑制了催化剂表面硫酸铵的形成,阻止了Ce的硫酸化,保护了催化活性位点。这与Nd改性CeO2/GO催化剂优异的抗SO2中毒能力相一致。
3 结论
通过一步浸渍法制备了Nd改性的CeO2/GO催化剂,用于NH3-SCR。与CeO2/GO相比,Nd0.5CeOx/GO在300 ℃时表现出优异的抗SO2中毒性能(在通入SO2气体4 h后NO转化率从91.4%下降到84.9%)。Nd和Ce均匀地分布在GO表面上,与GO形成了强相互作用,降低了GO的结晶度,从而有利于暴露更多的活性位点。并且用Nd改性CeO2/GO催化剂可以提高表面吸附氧的比值,有利于促进氧化还原反应。此外,GO为Nd0.5CeOx/GO催化剂提供了较大的比表面积,有利于反应气体的吸附。根据离子极化理论,Nd0.5CeOx/GO催化剂抗SO2中毒能力的增强应该归因于SO2更容易与Nd反应,形成相应的硫酸盐物种,从而减弱了Ce活性物种的硫酸化。
猜你喜欢
机电安全(2022年5期)2022-12-13
中华实验眼科杂志(2022年1期)2022-11-15
机械工业标准化与质量(2022年6期)2022-08-12
科学(2020年1期)2020-01-06
科教导刊(2016年36期)2017-03-30
中国调味品(2017年2期)2017-03-20
江苏农业科学(2015年11期)2016-01-27
中国卫生(2015年12期)2015-11-10
中国卫生(2015年10期)2015-11-10
中学化学(2015年2期)2015-06-05
