
早发型儿童失神癫痫的临床及EEG特征
2022-02-04 13:09:34聂健康盼云丁志杰李刚谢君伟王莉成林党小利
临床神经病学杂志 2022年6期
聂健,康盼云,丁志杰,李刚,谢君伟,王莉,成林,党小利
早发型儿童失神癫痫(EOAE)指起病年龄在3岁以前的失神性癫痫, 主要特征是典型失神发作、早期精神运动发育正常、大多数患者抗癫痫发作药物(ASMs)控制良好和远期预后良好。2017年国际抗癫痫联盟(ILAE)定义了四种具有典型失神发作的遗传性(特发性)全面性癫痫(GGEs):儿童失神癫痫(CAE)、青少年失神癫痫(JAE)、青少年肌阵挛癫痫(JME)和肌阵挛失神癫痫[1]。在研究[2]基础上,ILAE分类委员会于2005年提出了新的CAE诊断标准,指出CAE的发病年龄必须限制在4~10岁之间,高峰在5~7岁,而且神经功能发育正常,如有全面性强直-阵挛发作(GTCS)、肌阵挛发作等其他发作形式或棘慢波节律阵发短于4 s可作为排除标准。对于EOAE我国迄今尚无相关报导,国外文献中已有从几个月到3岁开始的典型失神发作儿童的报道[3-5]。一些学者[4]认为,婴儿期失神发作只是婴儿期其他GGEs综合征的一种发作形式,而不是一种独特的癫痫类型。据报道[5],在EOAE患儿中,多达12%的患者具有编码葡萄糖转运蛋白1型(GLUT1)的SLC2A1基因突变。本文旨在阐述EOAE的电-临床特征及其治疗转归,以期帮助更好地识别和治疗这类疾病。
1 临床资料
1.1 一般资料 研究对象系2012年8月至2021年12月天水市第三人民医院癫痫中心诊治的11例EOAE患儿(表1)。纳入标准:(1)出生至3岁内发作;(2)典型失神发作(持续数秒,少数10 s以上),频发(每天数十次),突发突止,意识障碍严重(意识丧失);(3)发作期EEG双侧对称同步3 Hz左右高波幅棘慢波节律性阵发,持续2~20 s;(4)伴或不伴有全面性强直-阵挛发作[6-7]。本组男孩6例,女孩5例;起病年龄均在3岁之内,平均起病年龄27个月。起病前所有患儿智力、运动发育均正常。其中两例患儿有一、二级癫痫家族史:病例3的奶奶患有癫痫(具体不详),病例6的爸爸在儿童期出现GTCS,成年后缓解。病例1和病例10在首次失神发作前有热性惊厥史。本组2例出生史异常:病例3为32周早产紧急剖宫产;病例9为臀位剖宫产。
1.2 临床表现 11例均有典型失神发作,其中5例以单纯性失神发作为唯一发作类型,表现为突然的意识丧失,正在进行的自主性活动和语言停止,双眼茫然凝视,表情呆滞,对外界刺激无反应,一般不伴跌倒或掉物。多数发作频繁,一日可达数次甚至几十次。发作持续数秒至数十秒后恢复,可继续发作前正在进行的动作。患者意识不到曾经历过发作,无发作后意识障碍,有些短暂的发作仅有一过性的轻微认知损伤,需非常仔细地观察才能发现。发作多发生在觉醒状态,睡眠期也可出现但不容易识别。其中4例失神发作合并有其他伴随症状,病例1伴有失张力成分,表现为发作时头部缓慢下垂,身体向后倾倒;病例2和病例5伴有口咽部自动症,与发作前正在进行的活动无关,表现为咂嘴、咀嚼、舔唇等简单动作;病例11一次发作中伴有节律性肌阵挛成分,表现为轻微的眼睑抽动;病例7和8病程中存在GTCS。
1.3 辅助检查
1.3.1 EEG特征 所有患儿均在我院完成至少2次19导联6 h视频EEG(VEEG)监测,包括完整的清醒-睡眠-清醒周期记录、间断节律性闪光刺激、过度换气诱发试验。同步体表EMG记录方法:分别在双侧三角肌、股四头肌和特殊抽动部位皮肤表面各放置1对盘状电极。所有EEG检查前进行睡眠剥夺均未使用镇静剂或催眠药。分析发作间期、发作期EEG及伴随症状和EMG改变的特点。11例患儿EEG背景活动均正常。6例出现间断一侧或双侧节律性后头部中-高波幅3~4 Hz左右θ活动(OIRDA)。4例患儿可见散发的限局性棘慢复合波(图1A),以额区最明显,也可出现在后头部。睡眠期较多2.5~3.5 Hz散发或阵发性不规则棘慢复合波片段(图1B),分布在双侧半球或限局在双侧额区。清醒期和睡眠中均可发作,主要在清醒期出现。能够完成过度换气的患儿均可诱发出典型失神发作,仅有1 例在14~20 Hz节律性闪光刺激下诱发失神发作或广泛性放电。发作期EEG为双侧对称同步的3 Hz(2.5~4 Hz)棘慢复合波节律爆发(图1C),波幅在放电过程中逐渐增高,并在接近结束时降低,棘慢复合波的频率在发作起始时稍快,平均为3.5~4.5 Hz,结束前频率稍慢,可达2.5~3 Hz,前头部波幅最高,持续 6~20 s,多数在 8~10 s左右,发作后背景活动无抑制或慢波现象。病例11伴有轻微的眼睑抽动,EMG呈短暂节律性肌电爆发,每次抽动对应的肌电爆发与广泛性棘慢波中棘波成分同步(图1D)。病例2和病例5失神发作后2~3 s伴有口咽部自动症(图1E),病例5在失神发作的后1/3时段出现手部自动症。病例6在3~4 Hz广泛性棘慢波爆发时,出现短暂(1.5~2 s)的失神发作(图1F)。
1.3.2 头颅影像学 11例患儿均进行了头颅影像学检查,除病例9 MRI扫描中检测到脑室周围白质T2加权信号强度增加外,其余均正常。
1.3.3 血尿代谢筛查与基因检测 均行血尿代谢筛查,采用血串联质谱+尿气相色谱分析法,11例患儿均无异常。其中4例进行家系全外显子检测,病例6发现SLC2A1基因的 c. 376 C>T(p.R126C)的杂合错义突变,经过家系分析此患儿父母在上述位点未发现异常。其余3例未发现致病基因及其他可能与本病相关的致病性突变。
1.4 治疗及随访 患儿在癫痫门诊随诊,复查VEEG;或通过电话询问方式获得近期预后情况。随访期22~204个月,平均71.6个月。本组11例均首选丙戊酸钠单药治疗,剂量为10~20 mg/(kg·d),在治疗中检测丙戊酸钠血药浓度,根据控制效果和血药浓度等调整药物剂量,6例于服药后2周至4个月发作完全控制。其中3例丙戊酸加足量后发作未控制,加服拉莫三嗪或氯硝西泮后发作完全控制。1例服用丙戊酸钠后患儿体重增加,改为拉莫三嗪,但服用2周后出现皮疹,给予停药,改用托吡酯口服后发作完全控制。病例4和病例5在丙戊酸钠完全控制下,分别在4.5~5.5岁时再次出现反复失神发作,复查VEEG仍有阵发性放电,继续服用丙戊酸后发作消失。病例11服用丙戊酸钠、左乙拉西坦、托吡酯治疗无效,仍然频繁发作和大量异常放电,给予了甲泼尼龙冲击治疗,方案为:甲泼尼龙冲击疗法。剂量为15~20 mg/(kg·d),1次/d,静脉输注,连用3 d为1疗程,4 d后重复第二疗程,连续2个疗程,之后改用泼尼松口服序贯治疗1.0~1.5 mg/(kg·d),足量服用1月后进入逐渐减量阶段,泼尼松总疗程2~3个月。糖皮质激素治疗的总疗程不少于3~5个月,具体时间取决于癫痫发作控制的程度以及EEG的转归情况。病例6服用丙戊酸、拉莫三嗪治疗无效,根据基因结果,给予生酮饮食治疗,1个月后发作明显减少,4个月至今再无发作。总体而言,目前所有患儿均无发作,其中5例在2~7年的总随访期内仍在接受治疗,其中6例不服用任何ASMs。
1.5 认知评估 由癫痫科医生和神经心理医生评估,9例患儿整体认知、语言和运动技能正常。携带SLC2A1基因突变的患儿认知功能轻度低下,1例患儿在学龄期学习困难合并有ADHD。

表1 11例EOAE患儿临床特征和治疗情况序号性别起病年龄家族史VEEG放电模式失神持续时间IPS/HV伴随症状或其他发作类型ASMs或其他治疗手段智力发育1男2岁8个月 FS+GSW、额区限局4~8 s-/+伴有失张力成分VPA+LTG正常2男1岁9个月-GSW15~18 s-/※伴有自动症VPA正常3女1岁12个月GGE(奶奶)GSW、后头部7~8 s-/※-VPA→LTG→TPM正常4男2岁6个月-GSW3~4 s-/※-VPA正常5男2岁7个月-GSW、不对称6~10 s-/+伴有自动症VPA正常6女11个月GGE(爸爸)GSW2~4 s-/※-VPA+LTG、生酮饮食偏低7女2岁9个月-GSW5~8 s-/+GTCSVPA正常8男1岁11个月-GSW、额区限局4~9 s-/※GTCSVPA+CZP正常9女2岁5个月-GSW7~8 s-/※-VPA+LTG正常10女1岁11个月FSGSW3~4 s-/※-VPA正常11男2岁1个月-GSW6~11 s+/+伴有眼睑肌阵挛成分VPA+LEV+TPM、激素学习困难、ADHD 注:FS+:热性惊厥附加症;FS:热性惊厥;GSW:广泛性棘慢波;VPA:丙戊酸钠;LTG:拉莫三嗪;※:不合作,无法完成;TPM:托吡酯;LEV:左乙拉西坦;ADHD:注意力缺陷多动障碍
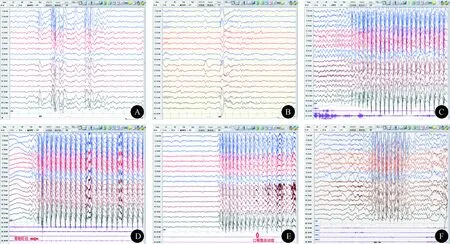
图1 早发型失神癫痫患儿发作期及发作间期的EEG特点
2 讨 论
EOAE被认为是GGEs疾病谱中的一种独特综合征,多有遗传性原因,其特征可描述为:从几个月到3岁之前起病,具有典型失神发作,伴或不伴有GTCS,男孩略占优势,早期精神运动发育正常,大多数患儿对ASMs的治疗反应良好,长期智力结果正常;但携带SLC2A1基因突变的EOAE的患者,癫痫的结局可能多变,一种药物完全控制或尽管采用多种治疗手段但仍然难以控制,并有轻度到重度的认知损伤。虽然表现与CAE相似,但具有不完全相同的电-临床特征、治疗反应和预后[7]。
典型CAE女性患儿发病率较高(60%~70%),与典型CAE相比,因本组病例偏少,EOAE患者的性别比例没有显著差异,男孩略占优势。本组近四分之一患儿的一、二级亲属有癫痫疾病史,他们均在儿童期起病,成年后可完全缓解。Chaix等[4]也观察到了这一现象,表明遗传易感性因素可能在EOAE的病因中起重要作用。本研究还发现本组中的个别患儿随着年龄的增长出现不同形式的GGEs或与年龄相关的放电模式,推测从幼儿到成年期GGEs之间存在病程的连续体。
失神发作是一种非惊厥性的癫痫发作,常突发突止,清醒期呆滞不动,伴意识丧失,持续3 s以上即可引起失神发作,EEG表现为双侧对称同步的3 Hz棘慢波爆发。本组中病例2和病例11发作持续时间较长,可长达10 s以上,但病例6在3~4 Hz广泛性棘慢波爆发时,出现短暂(1.5~2 s)的失神发作(图1F),即广泛性放电伴随着几乎难以察觉的意识丧失和失神发作。这种临床特征与Cavazzuti等[8]报道的病例相似,其描述了一名6个月大的婴儿患有失神发作,持续时间短至2 s。研究[4]表明,持续时间短暂的失神发作是早发型失神癫痫特征性的发作表现。
失神发作可有其他伴随症状,不同亚型的失神发作在预后方面无明显区别。病例11在失神发作过程中伴有节律性眼睑肌阵挛成分,发作时的眼睑EMG均显示与棘慢波的棘波成分呈锁时对应关系,并伴有光敏感性。眼睑肌阵挛癫痫(Jeavons综合征)通常发生在2~14岁之间的儿童,女孩多见。但一些研究[9]表明,这种类型的癫痫可在小于此年龄段的儿童中出现。因此应该注意到早发型失神发作患者伴有肌阵挛成分的问题。Chaix等[4]报导的1例EOAE患儿在服用拉莫三嗪后肌阵挛发作更加严重,考虑可能与服用拉莫三嗪有关系。拉莫三嗪对伴有肌阵挛成分的失神发作有加重作用已有报道[10]。Grosso等[11]研究表明,GTCS预测CAE的预后不良。本研究小组发现患者的临床结果与GTCS的存在之间没有相关性,对治疗的反应和总体预后也无影响。
限局性放电模式在特发性全面性癫痫的发作间期并不少见,本组病例1和病例8 VEEG记录到一侧或双侧额区限局性放电,病例3记录到双侧后头部限局性放电,但神经影像学无局灶性损伤,治疗和预后也无特殊。Lombroso等[12]在一个GGEs患者的大队列研究中,平均随访16年后发现,56%的患者在颞叶和额叶区域出现局灶性异常,在失神发作组中比例最高(60%)。Meencke等[13]报道了GGEs患者中发现微小皮质发育不良,EEG限局性放电可能起源于这些先天性的微小皮质发育不良。但是需要注意的是,虽然这3例患儿VEEG有限局性放电,但是以失神发作为唯一的发作类型,没有局灶性发作的特征,应避免诊断为局灶性癫痫。本组实验中过度换气诱发临床发作的阳性率不高,因部分患儿年龄偏小未能配合做诱发试验有关。失神发作的基本机制与丘脑-皮质环路的异常振荡节律有关,在丘脑-皮质环路中任何一点的放电都可能激活整个环路系统引起节律性振荡,其中丘脑的活动模式控制皮质棘慢波的节律。3岁之前的失神发作可能是由于产生3~4 Hz棘波复合波的特定丘脑-皮质环路不成熟所致[14]。失神癫痫通常遵循复杂的遗传规律,其中多个基因被认为是导致失神癫痫的原因之一。目前已知的EOAE的致病基因包括编码GABA受体亚单位(GABRG2)和钠通道亚单位基因(SCN1B)的基因突变[15-16]。最近发现的SLC2A1突变的表型可能与高外显率的常染色体显性遗传有关[17]。研究[15,18-19]表明,阵发性运动障碍、遗传性癫痫伴热性惊厥附加症(GEFS+)以及葡萄糖转运体1缺乏综合征(GLUT1-DS),都与早发型失神癫痫有关。Guerrini等[18]描述了6例患有失神癫痫合并阵发性运动障碍的儿童,起病年龄非常早(<3岁),临床症状和EEG特征表现一致,常规治疗对5例儿童无效。作者推测失神癫痫和阵发性运动障碍共存的患者,神经解剖中可能存在不同离子通道亚单位的参与。本组患儿均未出现阵发性运动障碍。
GEFS+是一种具有热性惊厥和多种非热性癫痫发作的综合征。Audenart等[15]报道了一个SCN1B基因突变的家系,先证者被诊断为早发型失神癫痫,其中热性惊厥和早发型失神癫痫同一个体中先后发生,失神发作不是由发热所引起,也从未出现过其他的发作类型。
有研究[17]表明,高达10%的EOAE患儿患有GLUT1-DS,由SLC2A1基因致病,为常染色体显性遗传。这类患者通常表现出除早发失神发作以外的其他神经特征,包括阵发性运动障碍、精神发育迟滞、面部畸形和ASMs反应差等。Suls等[20]已证明此类患者的额叶代谢永久性低下。进一步的研究应该评估SLC2A1基因筛查对失神癫痫和其他GGEs患者的诊断影响。生酮饮食对典型的GLUT1-DS效果明显,治疗后癫痫发作和EEG的异常放电显著减少。

关于EOAE是否是一种独特的癫痫综合征的争论仍在继续。本组认为EOAE是一种独特的癫痫综合征,其特征是从几个月到3岁之间起病的失神性癫痫,早期精神运动发育正常,大部分发作药物控制良好,长期智力发育正常,某些特征与典型的CAE重叠,但具有独特的特征。本研究发现EOAE伴有SLC2A1基因突变,其他特征有伴肌阵挛,失神发作时间短暂(<3 s),或EEG存在局灶性异常,即使发作得以控制,但会复发或遗留神经发育障碍等,总体预后似乎不如CAE。EOAE并不常见,多中心研究将有助于归纳诊断要点和预后特征。
猜你喜欢
中国民间疗法(2021年5期)2021-06-09 09:21:04
北方文学(2020年6期)2020-06-19 08:02:20
婚育与健康(2020年12期)2020-02-26 01:51:39
饮食科学(2017年5期)2017-05-20 17:11:53
广西林业科学(2016年3期)2016-03-16 05:43:23
医学研究杂志(2015年5期)2015-06-10 06:43:26
西南军医(2015年4期)2015-01-23 01:19:30
中国中医药现代远程教育(2014年20期)2014-03-01 04:31:21
四川生理科学杂志(2014年1期)2014-02-28 14:08:24
保健与生活(2012年5期)2012-06-04 23:07:01
