
Comparative profiling of immune genes improves the prognoses of lower grade gliomas
2022-02-04 08:21ZhiliangWangWenChengZhengZhaoZhengWangChuanbaoZhangGuanzhangLiAnhuaWuTaoJiang
Cancer Biology & Medicine 2022年4期
Zhiliang Wang*, Wen Cheng*, Zheng Zhao, Zheng Wang, Chuanbao Zhang, Guanzhang Li, Anhua Wu,Tao Jiang,
1Department of Neurosurgery, Beijing Neurosurgical Institute, Capital Medical University, Beijing 100050, China; 2Department of Neurosurgery, The First Hospital of China Medical University, Shenyang 110001, China; 3Department of Neurosurgery,Beijing Tiantan Hospital, Capital Medical University, Beijing 100050, China
ABSTRACT Objective: Lower grade gliomas (LGGs), classified as World Health Organization (WHO) grade II and grade III gliomas, comprise a heterogeneous group with a median survival time ranging from 4–13 years. Accurate prediction of the survival times of LGGs remains a major challenge in clinical practice.
KEYWORDS Lower grade glioma; immune; gene pairs; signature; prognosis
Introduction
Glioma is the most common and lethal brain tumor in the central nervous system1. According to the World Health Organization (WHO) grading system, gliomas are classified into grades I–IV based on their histological characteristics2.Recent studies have described grade II and III gliomas as lower grade gliomas (LGGs)3-5, which are less aggressive than grade IV glioblastomas (GBMs). Patients with LGG are recommended to receive surgical resection combined with radioand/or chemotherapy. However, due to its invasive nature,complete resection of LGGs is almost impossible, and local recurrence occurs at variable intervals. In addition, a considerable subset of LGGs progress to GBM within months, while others remain stable for years. Prediction of LGG survival,ranging from 1–15 years, still remains a major challenge. The identification of subsets of patients at high risk for recurrence and death, who may benefit from additional systemic therapy,is therefore urgently needed.
In 2016, the WHO developed a glioma classification system based on the integration of multiple genotypic events(IDH mutations and 1p/19q co-deletions), highlighting the prognostic roles of specific molecular parameters. A growing number of studies have proposed gene expression signatures for survival stratification of patients with LGG. However,none of them has been incorporated into clinical practice due to numerous issues, including overfitting on small discovery datasets and lack of clinical validation. The clinical application of approaches based on gene expression is also hampered by the heterogeneities between datasets and technical bias due to the use of multiple measurement platforms. To develop a robust signature with higher chances of clinical application,novel methods are therefore required for data processing.
Immunotherapy is a promising treatment in multiple cancers, including gliomas. Rather than time-honored “immune privilege”, it is now clear that immunological disorders are involved in initiation and progression of gliomas. We previously reported that excessive immune response and disorganized immune microenvironment strongly contributed to the short survival of glioma patients6-10. In addition, the validity of prognostic prediction has been significantly enhanced by integrating clinicopathological and immunological features11.In this study, we reviewed the transcriptomic data of 865 LGGs to develop and validate a signature based on the differential profiles of immune gene pairs (IGPs). An individualized model integrating these immune signatures with clinical characteristics was constructed, to achieve an improved estimate of survival times in patients with LGG.
Material and methods
Patients
In this multiple cohort study, transcriptomics analysis was applied to LGG patients from 4 independent cohorts(Figure 1A). The expression data were collected from the Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn/index.jsp), The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/) databases, and the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse16011). A total of 865 LGG patients were analyzed (Figure 1B). The in-house CGGA RNAseq and microarray cohorts were used for training and internal validation, respectively, and TCGA RNAseq and GSE16011 cohorts were analyzed for external validation. This study was approved by the Medical Ethics Committee of Beijing Tiantan Hospital(Approval No. KY2014-002-02).
Tissue samples and molecular testing
The CGGA patients were treated by members of the CGGA group. Tumor tissue samples were collected at the time of surgery after obtaining informed consent. Neuropathologists established the diagnoses and ensured the quality of tissues for molecular testing. Overall survival (OS) was calculated from the date of diagnosis until death or the end of follow-up. The point of death was defined by death certification, which was obtained from local hospitals or police stations.
The tissue samples were immediately snap-frozen in liquid nitrogen after surgery. The percentage of tumor cells was assessed by hematoxylin and eosin staining, and the samples with more than 80% tumor cells were selected for RNA extraction. Total RNA from tumor samples was extracted by using the RNAprep pure Tissue Kit (Tiangen, Beijing, China)according to the manufacturer’s protocols. The concentration and optical density of RNA were measured using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies,Wilmington, DE, USA).
The IDH1/2 mutation status was determined by pyrosequencing, as previously described12. Loss of 1p and 19q chromosome arms was inferred with a Gaussian window smoothing algorithm from RNA sequencing data, and exhibited a good concordance with the 1p/19q status determined by the SNP array (Mathews correlation coefficient = 0.94,P<0.0001)13.
Immune gene selection
A total of 2,879 immune-related genes were collected from the ImmPort database (https://immport.niaid.nih.gov) and Gene Set Enrichment Analysis (GSEA) dataset (http://software.broadinstitute.org/gsea/index.jsp). Among them, 2,214 immunological genes shared by three transcriptomics cohorts were reserved for further analyses (Figure 1C). The immunological genes from the ImmPort database were functionally categorized into 10 subsets (antigen processing and presentation,antimicrobials, B cell receptor signaling pathway, chemokines,chemokine receptors, cytokines, cytokine receptors, natural killer cell cytotoxicity, and the T cell receptor signaling pathway) (Figure 1D).
Functional annotation and analysis
DAVID Bioinformatics Resources, version 6.8 (https://david.ncifcrf.gov/) was used to provide a comprehensive understanding of gene functions and biological processes. A false discovery rate less than 0.05 was regarded as statistically significant.

Figure 1 Continued
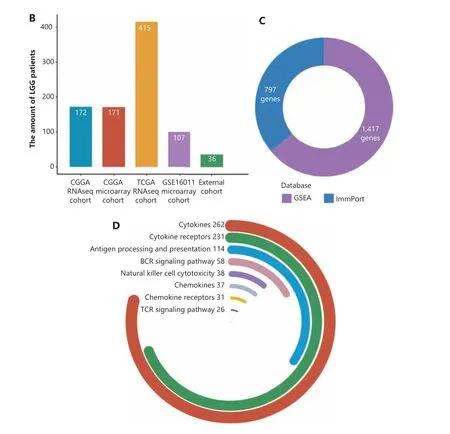
Figure 1 The study design. (A) Workflow graph of this study. (B) The histogram shows the number of patients collected from 4 cohorts.(C) A total of 1,417 and 797 immune-related genes were collected from the Gene Set Enrichment Analysis and ImmPort databases, respectively. (D) The immune-related genes from the ImmPort database were stratified into 7 categories, and the genes for cytokines, cytokine receptors, and antigen processing and presentation groups comprised the highest percentages of immune genes.
Assessment of immune cell infiltration
The Microenvironment Cell Populations-counter method14and CIBERSORT algorithm with LM22 gene signature, with 2 deconvolution methods, were used to provide highly precise quantitative information on the tumor microenvironment (TME) of cell contents in heterogeneous tissues, which allowed for sensitive and specific discrimination of the human immune cell and stromal cell phenotypes from transcriptome data, including B cells, T cells, natural killer cells, macrophages, dendritic cells, myeloid subsets, endothelial cells,and fibroblasts.
Development of IGPs
Qualitative assessment is generally more reliable than quantitative assessment in differential gene expression analysis.Before developing the gene pairs, we arranged genes in the order of the initial letter. The IGPs were analyzed by pairwise comparisons of immune genes based on their expression values. Each IGP was viewed as an independent event with two possible outcomes [Gene(i) expression > Gene(j) expression or Gene(i) expression <Gene(j) expression]. Briefly,patients with the IGPs [Gene(i) expression > Gene(j) expression] were given a score of 1. Patients with IGPs [Gene(i)expression <Gene(j) expression] were given a score of 0. This was repeated for every immune gene pair to generate an IGP score for each patient.
Real-time PCR (RT-PCR)
A total of 36 samples, including 13 WHO grade II gliomas (8 1p/19q intact and 5 co-deletion patients) and 23 WHO grade III gliomas (13 1p/19q intact and 10 co-deletion patients),were collected to assess the prediction accuracy and clinical usefulness of the nomogram model by real-time quantitative PCR. Specifically, 1 µg of total RNA was reverse-transcribed into cDNA using a RevertAid First Strand cDNA Synthesis Kit(Thermo Fisher Scientific, Waltham, MA, USA). The results of RT-PCR were normalized to the corresponding glyceraldehyde 3-phosphate dehydrogenase mRNA levels and the analyses were performed in triplicate to remove the outliers. Relative gene expression was determined using the 2-dCtmethod. The primers were as follows:
CRH: forward: 5′-GGGAACCTCAACAAGAGCCC-3′, reverse:5′-AACACGCGGAAAAAGTTGGC-3′
IFNB1: forward: 5′-GCTTGGATTCCTACAAAGAAGCA-3′,reverse: 5′-ATAGATGGTCAATGCGGCGTC-3′
HOXA9: forward: 5′-AAAAACAACCCAGCGAAGGC-3′,reverse: 5′-ACCGCTTTTTCCGAGTGGAG-3′
PRG3: forward: 5′-CAACTATCGCATTCAGTGCTGC-3′,reverse: 5′-GGGACCAGTAAGCAAAATTCCA-3′
IL10: forward: 5′-TCAAGGCGCATGTGAACTCC-3′, reverse:5′-GATGTCAAACTCACTCATGGCT-3′
IL9: forward: 5′-CTCTGTTTGGGCATTCCCTCT-3′, reverse:5′-GGGTATCTTGTTTGCATGGTGG-3′
PTH2: forward: 5′-GTAGGGGACTGTGCGGGAAG-3′,reverse: 5′-CTCCATCACCTGTGGAGAACC-3′
RETNLB: forward: 5′-AGCTCTCGTGTGCTAGTGTC-3′,reverse: 5′-TGAACATCCCACGAACCACA-3′
NKX2-5: FORWARD: 5′-CAAGTGTGCGTCTGCCTTTC-3′,reverse: 5′-CGCACAGCTCTTTCTTTTCGG-3′
PRLH: forward: 5′-TGCAAGTCGTACCCATCGG-3′, reverse:5′-GGCGTACCAGGCAGGATTG-3′
NKX3-2: forward: 5′-ACCGAGACGCAGGTGAAAAT-3′,reverse: 5′-CACCTTTACGGCCACCTTCT-3′
UCN3: forward: 5′-GAGGCACCCGGTACAGATAC-3′,reverse: 5′-GAGGGACAGGGTGAACTTGG-3′
NR2C1: forward: 5′-CCAGATTGTGACAGCACTTGA-3′,reverse: 5′-CTTGGAGTAGAGCCGTCGT-3′
PRLHR: forward: 5′-TGAGTTCGGCCTGCTACAAC-3′,reverse: 5′-CCTGGCTAAGTGGCATCAGA-3′
REG1A: forward: 5′-ACCGGACCATCTCTCCAACT-3′,reverse: 5′-AGGGTTCCAAAGACTGGGGT-3′
TRIM31: forward: 5′-CGCAATCAGGTTCAACTCGC-3′,reverse: 5′-CTCGGGCATGTAGCCTCTTT-3′
PTX3: forward: 5′-CGAAATAGACAATGGACTCCATCC-3′,reverse: 5′-CTCATCTGCGAGTTCTCCAGCA-3.′
Statistical analysis
All figures and statistical analyses were performed based on R for Windows, version 3.4.2 (http://www.r-project.org).The least absolute shrinkage and selection operator method from the glnmet package was used to reduce the overfitting.The risk-score formula for predicting survival was developed based on the 10 IGPs and the regression coefficient derived from Lasso regression analysis. The risk score for each patient was calculated as follows:
Risk score = (β1×score(IGP1)) + (β2×score(IGP2)) + (β3×s core(IGP3)) + (β4×score(IGP4)) + (β5×score(IGP5)) + (β6×s core(IGP6)) + (β7×score(IGP7)) + (β8×score(IGP8)) + (β9×s core(IGP9)) + (β10×score(IGP10))
Standard median splits can be used on either continuous or ordinal variables to convert them into dichotomous variables15,16. In the present study, patients were separated into low and high risk groups based on the cut-off point (median value). The Kaplan-Meier survival curve and log-rank test were used to evaluate the differences in OS. Univariate and multivariate Cox proportional hazard models were built using the Cox proportional hazards function from the survival package. All independent prognostic factors using multivariate analyses were then assessed by using the nomogram. The bootstrap method (B= 1,000) was performed to calculate the concordance index (C-index) for each independent dataset.P<0.05 was considered as statistically significant.
Results
Identification of 402 prognostic IGPs
To identify the IGPs for model development, the following steps were performed. First, the 2,214 shared immune genes were examined by pairwise comparisons to generate 2,449,791 IGPs. Next, we excluded unevenly distributed IGPs (displaying over 95% of the scores equal to 1 or 0) from the 3 datasets.Then, a total of 15,957 overlapping IGPs were identified for further analyses. The prognostic values of the IGPs were evaluated using the log-rank test. There were 4,464, 4,911, and 3,557 prognostic IGPs in the CGGA RNAseq cohort (training),CGGA microarray cohort (internal validation), and TCGA RNAseq cohort (external validation), respectively. We found that there was a modest overlap of prognostic IGPs (n= 411)between these 3 cohorts. After removal of 9 controversial gene pairs, we obtained a list of 402 prognostic IGPs.
Functional analysis of IGPs
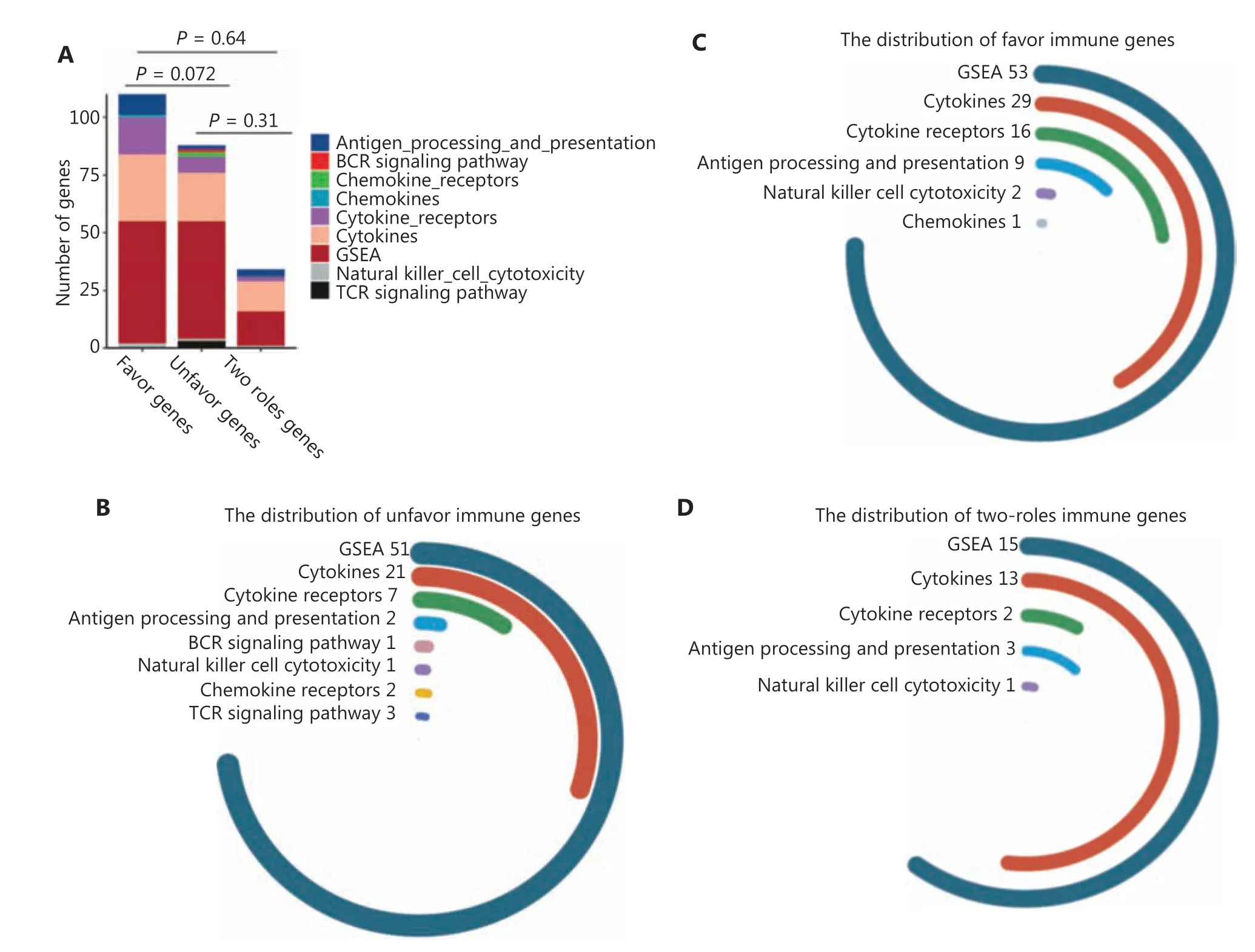
Figure 2 Continued
To verify the prognostic utility of the selected IGPs, we defined IGPs with HR > 1 as unfavorable prognostic predictors (UPPs,n= 277), and the others as favorable prognostic predictors(FPPs,n= 125). The genes with higher expressions among UPPs or with lower expression among FPPs were identified as unfavorable genes (UGs,n= 88). In turn, the other genes were defined as favorable genes (FGs,n= 110). We found that FGs comprised a larger percentage of cytokine, cytokine receptor,and antigen processing genes (P= 0.072). Moreover, 34 genes behaving as both FG and UG among prognostic IGPs were termed two-side genes (TSGs) (Figure 2A). Figure 2B and 2D shows that FGs, UGs, and TSGs mainly included genes for cytokines, cytokine receptors, and proteins involved in antigen processing and presentation, as well as in natural killer cell cytotoxicity.
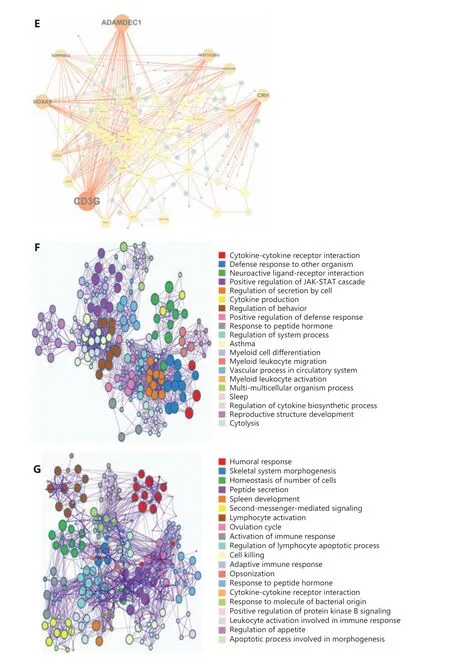
Figure 2 The landscape of 402 immune-related gene pairs (IPGs). (A) The 402 IPGs were composed of 110 favorable genes (FGs), 88 unfavorable genes (UGs), and 34, 2 role genes. (B–D) The distribution of immune genes from the GSEA and ImmPort databases for the FGs, UGs,and 2 role genes. (E) The gene co-expression network comprised of 232 unique immune genes in 402 immune-related gene pairs. The CD3G,ADAMDEC1, HOXA9, HIST1H2BG, CCR4, CRH, and SERPINB12 genes showed the highest connection degrees in the network. Biological function analyses of FGs (F) and UGs (G).
A gene co-expression network was constructed to evaluate the roles of IGPs. Figure 2E shows that the IGP co- expression network was represented by 232 unique immune genes.Of these, 7 genes with the highest connection degree wereCD3G,ADAMDEC1,HOXA9,HIST1H2BG,CCR4,CRH, andSERPINB12(more than 15 connections/interactions). Among them,CD3G(n= 50),ADAMDEC1(n= 37),HOXA9(n= 28),HIST1H2BG (n= 20), andCCR4(n= 16) were core members of the UPP subset, accounting for 54.5% of the total 277 UPPs. However,CRH(n= 27) andSERPINB12(n= 18) were deemed as the core genes of the FPP subset. Further examination of the role of the above core genes suggested thatCD3G, ADAMDEC1,HOXA9,HIST1H2BG, andCCR4were significantly overexpressed in GBM and associated with unfavorable prognoses, while theCRHhad a protective function(Supplementary Figure S1).
We then used GO analysis to characterize the biological and functional annotations of the FGs and UGs. Because these subsets were related to immune functions, there were both similarities and differences with their GO annotations. Figure 2F shows that all FGs and UGs were significantly correlated with cytokine-cytokine receptor interactions and with responses to peptide hormones. In addition, FGs were strongly enriched in functions related to myeloid cell differentiation, myeloid leukocyte migration, myeloid leukocyte activation, and positive regulation of the JAK-STAT cascade. However, the UGs were mainly involved in lymphocyte regulation and physiological functions, including second-messenger-mediated signaling,skeletal system morphogenesis, and regulation of appetite(Figure 2G).
The comparative score of core immune genes was a prognostic factor
To determine the prognostic accuracy of the differential expression profiles, we developed an HR scoring algorithm to calculate the comparative pattern of specific genes. TheHOXA9-related IGPs were the third highest number of unfavorable gene pairs in 402 prognostic IGPs. There was a total of 28 HOXA9-related IGPs. The HR score ofHOXA9-related IGPs ranged from 0–28 for each patient. Figure 3A shows that the majority of LGG patients had aHOXA9-score of 0. Survival analysis revealed that these patients had a better prognosis than those with aHOXA9-score of 28 (Figure 3B,P= 0.00015).When strata were considered based on the 0–14 score cohorts and 15–28 score cohorts, patients in the higherHOXA9-score cohorts exhibited a significantly reduced OS compared to those in the lower HOXA9-score cohorts (Figure 3C,P<0.0001).Furthermore, when patients were divided into 5 groups based the HOXA9-score, patient survival tended to decrease with an increase in theHOXA9-score (Figure 3D,P<0.0001).
TheCRH-related IGPs were the most frequently favorable gene pairs in 402 prognostic IGPs. There was a total of 27CRH-related IGPs. The HR score ofCRH-related IGPs ranged from 0–27 for each patient. Similar to theHOXA9-RGPs prediction model, risk prediction on the FPPCRH-score resulted in a significant stratification of OS. Specifically, patients with aCRH-score of 27 had a better prognosis than those with aCRHscore of 0 (Figure 3E and 3F,P<0.0001). When patients were classified into 2 or 5 groups based on theCRH-score, a robust inverse correlation was observed between theCRH-score and survival time (Figure 3G and 3H). These results showed that the gene pair patterns of core immune genes enhanced our understanding of gene relationships and provided valuable information for the management of glioma patients.
Prognostic significance of the immune signature in the discovery and validation cohorts
To generate an immune signature with prognostic value,we conducted LASSO regression based on the 402 prognostic IGPs to minimize the risk of overfitting (Figure 4A and 4B). Ten IGPs, including 4 UPPs (HOXA9-PRG3,NKX2-5-PRLH, IL10-IL9,NKX3-2-UCN3) and 6 FPPs (CRH-IFNB1,IL9-PTH2,IL9-RETNLB,NR2C1-PTX3,PRLHR-REG1A, andPRLHR-TRIM31), were selected using LASSO regression analysis (Figure 4C).
To determine the prognostic value of the immune signature,the median risk value was defined as the cutoff for patient classification into high or low risk groups. We found that patients in the low risk group (median survival: undefined) had a significantly longer survival time than patients in the high risk group [median survival: 37.76 months; hazard ratio (HR),7.58; 95% confidence interval (CI), 4.31–13.31,P<0.0001](Figure 4D). The same formula was used for the internal and external validation cohorts to further verify the prognostic utility of the immune signature. Patients were also divided into low and high risk groups according to the cutoff value. As expected, patients in the low risk group had a longer survival time than patients in the high risk group (CGGA microarray cohort, median survival, undefinedvs.45.8 months. HR,7.02; 95% CI, 4.04–12.22,P<0.0001; TCGA cohort, median survival, 134.18 monthsvs.62.91 months; HR, 3.51; 95% CI,2.26–5.48,P<0.0001) (Figure 4E and 4F). The results indicated that the identified immune signature had robust prognostic value across different testing platforms.

Figure 3 The accuracy of the hazard ratio (HR) scoring model in prognosis predictions. (A) The composition of different subgroups defined by the HR scores of HOXA9-RGs. (B) The Kaplan-Meier estimate showed the survival curves of patients in HOXA9-RGs in the 0 score and 28 score groups. (C) Similar to panel B, the Kaplan-Meier estimate showed the survival curves of patients in the low HOXA9-RGs score group(scores of 0−4) and high HOXA9-RGs score group (scores of 15−8). (D) The HR scores of HOXA9-RGs were separated by every 10 scores plus the minimum and maximum scores. The Kaplan-Meier curves of patients in 5 scored cohorts. (E–H) The CRH-RGs scoring model for predicting the overall survival of lower grade gliomas.
Next, patients were stratified based on clinicopathological features, including WHO grade (II and III), 1p/19q status (co-deletion and intact), IDH status (mutation and wild-type), and histology (astrocytoma and oligoastrocytoma). In the CGGA RNAseq cohort, the immune signature was an unfavorable indicator in all stratified analyses(Supplementary Figure S2) and patients in the low risk group presented a better prognosis than those in the high risk group.Furthermore, the high risk group tended to present a poor outcome in subgroups of the CGGA microarray (Supplementary Figure S3) and TCGA RNAseq (Supplementary Figure S4)cohorts, even if some of the results were statistically marginally significant.
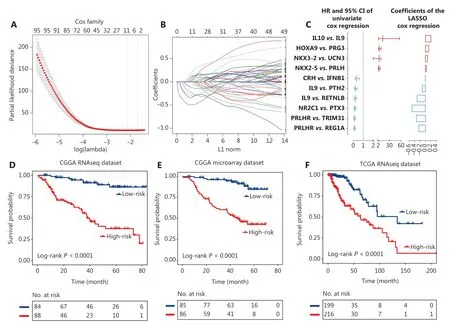
Figure 4 Identification of the 10 immune-related gene pair (IGP) signature for lower grade glioma patients. (A) One-thousand times cross-validation for tuning parameter selection in the LASSO Cox regression model. (B) LASSO coefficient profiles of 10 IGPs by the largest value of lambda (1se). (C) The hazard ratios and the 95% confidence intervals using univariate analyses, and the values of coefficients using Lasso analyses of 10 IGPs. The dichotomized immune signature risk score allowed the segmentation of patients into high and low risk groups in the Chinese Glioma Genome Atlas (CGGA) RNAseq cohort (D), CGGA microarray cohort (E), and The Cancer Genome Atlas RNAseq cohort (F).
In addition, uni- and multivariate Cox regression analyses revealed that the immune signature was an independent prognostic factor for LGG patients in 3 independent cohorts after adjusting for age, sex, WHO grade, IDH status, and 1p/19q status (Supplementary Table S1). Moreover, HR analysis showed that the presence of the immune signature was a significant detrimental factor in nearly all subgroups (Supplementary Figure S5). Therefore, the immune signature was a robust predictor of OS in LGG patients.
Association of the immune signature with tumor immune infiltration
To characterize the immune risk score, we analyzed the association between the risk score and clinico-pathological and tumor-related immune parameters. The TME composition,composed of glioma purity, and human immune cells and stromal cells, was deconvoluted using the ESIMATE method,MCPcounter, and CIBERSORT algorithm. The glioma samples were arranged in order of increasing risk scores (Figure 5A).Pearson’s correlation analysis showed that the risk score had a significant positive association with the immune and stromal scores. Patients in the high immune risk score group mainly exhibited higher infiltration of M2-polarized TAMs and T cell CD4 resting memory. The infiltration of naïve B cells, activated mast cells, and plasma cells were significantly higher in the lower immune risk group (Figure 5B). In terms of clinical characteristics, we found that grade II glioma patients, as well as patients with the IDH mutation, 1p/19q codel, and methylated MGMT promoter were more likely to be in the low immune risk score group (Figure 5A).

Figure 5 Continued

Figure 5 The association between immune signature and tumor immune infiltration characteristics. (A) The landscape of clinical, molecular features, and tumor microenvironment (TME) cells together with immune risk score (a, The association between risk score and continuous variables was assessed using Pearson’s correlation tests. b, The distribution of risk score between 2 groups was assessed using one-way analysis of variance). (B) The distribution characteristics of TME cells between low and high immune risk score groups. The scattered dots represent TME cell values and the thick lines represent the median value (*,**, ***, and **** represent P <0.05, P <0.01, P <0.001, and P <0.0001,respectively). (C) The normalized expression value of immune activation-relevant genes in the 2 groups. (D) The normalized expression values of immune checkpoint-relevant genes in the 2 groups. (E) The relationship between immune risk scores and T-cell-related metagenes.
Moreover, the cytokine and chemokine environments characterizing the low and high immune risk score groups were analyzed17. TheIDO1,CD274,HAVCR2,PDCD1,CTLA4,LAG3, andPD-L2genes were identified as immune checkpoint-relevant transcripts9(Figure 5C). TheCXCL10,CXCL9,GZMA,GZMB,PRF1,CD8A,IFNG,andTBX2genes were identified as immune activation-related transcripts(Figure 5D). The high immune risk score group exhibited a higher expression of immune checkpoint-related genes, while the immune activation-related genes displayed a relatively poor expression in the low immune risk score group. To gain a deeper understanding of the relationships between inflammation and the immune risk scores, 7 lymphocyte-specific metagenes, including 7 subtypes of immune inflammation response genes, were collected18. Figure 5E shows that the activities ofMHC I,MHC II, andSTAT1were specifically elevated in the high immune risk score group. Together, the results indicated that gliomas with higher immune risk scores were more likely to possess a more complex physiological immune homeostasis.
Development and validation of a prognostic nomogram
To maximize the predictive accuracy, a nomogram combining our immune signature with traditional clinical features was developed. Three independent covariates (immune signature,1p/19q status, and grade) were selected using multivariate Cox regression analysis (Figure 6A). The nomogram was able to predict OS with a c-index of 0.85, which was significantly higher than those obtained with the individual traditional clinical and the combined traditional clinical factors (Figure 6B).The calibration plots indicated an optimal agreement between the predictions and the observed 1, 2, 3, and 5-year survival percentages (Figure 6C).
To test the universality of the identified immune signature with respect to other populations and platforms, the nomogram was applied to validation cohorts, yielding c-indices of 0.80 in the CGGA microarray cohort (internal validation),0.80 in TCGA RNAseq cohort (external validation), and 0.79 in the GSE16011 microarray cohort (independent validation).The predicted survival times based on the nomogram were highly consistent with the actual survival data from the validation tests (Figure 6D, 6F and Supplementary Figure S6).
We also collected independent tumor tissue samples, including 36 LGG patients, for immune signature validation using RT-PCR (Figure 6G). The nomogram confirmed its predictive robustness in this cohort, with a c-index of 0.75 (Figure 6H).These results showed that the nomogram was accurate in predicting patient survival, and proved to be a new potential tool for use in clinical practice.
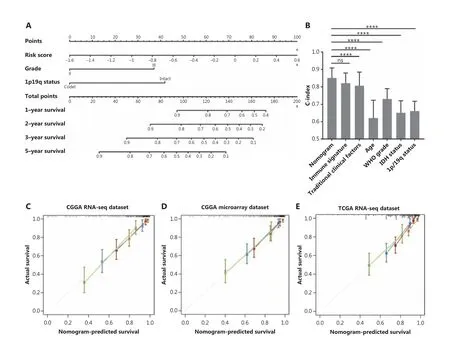
Figure 6 Continued

Figure 6 The performance of the nomogram in the training and validation cohorts. (A) The nomogram for predicting 1-year, 2-year, 3-year,and 5-year overall survivals for lower grad glioma (LGG) patients. (B) The c-index in predicting overall survival (OS) was compared between the nomogram model and other factors, including immune signature, age, World Health Organization grade, IDH status, and 1p/19q status in the Chinese Glioma Genome Atlas RNAseq cohort (mean ± SD; ****P <0.0001, Student’s t-test). The calibration curve for predicting the OS for LGG patients in training (C), internal validation (D). and external validation (E–F); goldenrod: 1-year survival, firebrick: 2-year survival, steel blue:3-year survival, and dark olive green: 5-year survival. (G) The heat map shows the 10 immune-related gene pair profiles with clinicopathology information of 36 frozen tissue LGG samples. (H) The calibration curve for predicting the OS for the 36 patients.
Discussion
Accounting for approximately one-third of adult gliomas,LGGs show highly variable clinical behaviors. Subgroups of LGG exhibit poor outcomes, similar to GBM, in spite of substantial differences in histology and genetic background19.Discrimination of patients at high risk of death, who might benefit from additional intensive treatment, remains one of the major clinical challenges. Although great effort has been expended in the identification of gene expression-based prognostic signatures, their clinical application is still limited.
In this study, we reviewed gene expression profiles from different populations and designed a robust transcriptomic comparison method based on microarray, RNAseq, and routine qPCR technologies. We characterized the prognostic profiles of gene pairs, which resulted in an immune signature that allowed for improved estimation of LGG patient survival.Conventional methods based on differentially-expressed genes are of limited applicability due to differences of the sequencing platform and batch effects. The gene-pair comparison method minimizes the impact of the batch effect, using differentially-expressed genes at the individual level.
It has been well-established that innate and adaptive immune systems promote glioma malignancy by multiple mechanisms20-22. Recent studies proposed immunotherapy as a promising strategy for glioma23-25. In comparison with GBM, LGG displays moderate immune-mediated reactions characterized by weakened local immune response and lower immune cell infiltration26. We previously reported an immune gene signature for GBM, suggesting that the estimation of immune genes was a valuable strategy for prognosis and prediction of treatment effectiveness27. However, few immunological signatures have been established for LGG. Here, an immune signature was established based on pair-wise comparisons. Unlike traditional methods for the normalization of gene expression data, this approach was based on comparative gene expressions, which provided more reliable results, avoiding the heterogeneity between data sets and technical bias related to the diversity of measurement platforms. Therefore,this signature exhibited a robust predictive performance across different platforms and populations.
A total of 402 IGPs, including 277 UPPs and 125 FPPs,exhibited high prognostic consistency for LGG across 3 distinct datasets. The GO analyses revealed that FG-related immune functions, such as myeloid leukocyte differentiation, activation, and migration were closely correlated with the promotion of an anti-tumor immune responses and with a favorable prognosis for LGG patients.HOXA9andCRHwere core genes, which were more frequent among the IGPs. HOXA9 is a homeodomain-containing transcription factor, predicting poor survival in patients with leukemia28, ovarian cancer29and breast cancer30. CRH is a peptide hormone involved in the response of stress and inflammation31,32. The role of these factors in other cancers is consistent with our findings, which further confirmed the potential suitability ofHOXA9andCRHstatus as predictors of clinical outcome. Moreover, our HR scoring system proved an excellent predictor of survival,providing a model for risk stratification based on core gene comparative profiles.
In this study, an immune signature was developed based on the profiling of 402 IGPs. After reducing the overfitting of the gene pairs profile by the LASSO regression model, a risk signature for glioma was developed, based on 10 comparative gene pairs. IL10 was the one of the key interleukins composed of unfavorable IGPs. Interleukin 10 encoded by theIL10gene, is an anti-inflammation cytokine33. IL10 is involved in ERK1/2,p38, and NF-κB signaling, and down-regulates co-stimulatory molecules on macrophages34. Macrophages are key cells in the innate immune system. They phagocytose pathogens and cellular debris, promote inflammation, and have important roles in tumor immunity35-37. Depending on the microenvironment, macrophages can polarize to M1 (inflammatory) or M2(anti-inflammatory) phenotypes. Rafael et al.38,39found that the IL-10/IL-10R axis was required for polarization of microglia to the M2-like phenotype, and promoteed tumor growth in an IL-10-dependent manner. In addition, M2-polarized TAMs promoted tumorigenesis, the epithelial-mesenchymal transition, proliferation, and infiltration through the IL10-related signaling pathway in cancers40-42.
Because of the robust relationship between genes, the immune signature retained its prognostic significance in 3 large LGG cohorts tested by RNAseq or microarray platforms.Furthermore, the immune signature, either as a continuous or categorical variable, was an independent prognostic factor,after adjustment for clinical and molecular characteristics.These findings highlighted the potential of the immune signature as a new tool to improve prognostic accuracy.
Based on the analysis of the relationship between the immune signature and the TME, we found that patients in the high risk group had a high number of specific immune cell populations (T cells, CD8+ T cells, cytolytic lymphocytes, and monocytic lineage), and exhibited elevated expressions of genes involved in T-cell activation (CXCL10,CXCL9,GZMA,GZMB,PRF1, andTBX2), antigen presentation (MHC IandMHC II), and interferon signal transduction (STAT1). In addition,the immune checkpoints (PDCD1,CD274,PD-L2,HAVCR2,CTLA4,LAG3, andIDO1), which block T cell activation, were significantly upregulated in patients from the high risk group.This observation indicated that patients with a high immune risk score tended to have more complex interactions between gliomas and their immunological microenvironments.
Although numerous valuable prognostic factors are known,the appropriate combination of markers for individualized prognostic predictions is still needed. Nomograms are graphical depictions of predictive statistical models, with a proven advantage over traditional systems for individualized prediction, and have been developed for various types of cancers43.To date, the assessment of nomograms has been rarely conducted for LGG patients. Here, we developed a nomogram prediction module by integrating the identified immune signatures with routine clinical features to predict 1-year, 2-year,3-year, and 5-year survival in LGG patients. The nomogram module exhibited more accurate projections in predicting OS than other prognosis markers reported in previous studies44.
We recognize some limitations in this study. First, some important molecular markers, including TERT, MGMT,CDKN2A/B, were not collected from TCGA, CGGA, and GSE16011 cohorts. Second, the GSE16011 and TCGA cohorts did not provide information of the extents of surgical resections. Third, there was some missing data of the 1p/19q status and IDH status in the GSE16011 cohort.
To our knowledge, this nomogram was the first to apply a gene comparison approach to gliomas, and exhibited excellent predictive accuracy across RNAseq, microarray, and qPCR cohorts. The nomogram provided an individualized and comprehensive estimate of the prognostic risks and may substantially contribute to clinical management decisions.
Conclusions
In the present study, we first profiled the comparative pattern of immune genes and identified an immune signature based on 10 gene pairs with independent prognostic values of LGG patients. We established an individualized prognostic model combining the immune signatures with clinical information.Our study possessed good versatility and efficiency, large RNA sequencing cohorts for discovery and validation, and a robust validity based on RT-PCR. However, our study was limited due to its retrospective nature and should be validated by future studies. Overall, our findings provided clues to estimate the survival of LGG patients, and has great promise for the identification of novel molecular targets.
Grant support
This work was supported by grants from the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (Grant No. ZYLX201708), the National Natural Science Foundation of China (NSFC)/Research Grants Council (RGC) Joint Research Scheme (Grant No. 81761168038), the Beijing Municipal Administration of Hospitals’ Mission Plan (Grant No. SML20180501), the National Natural Science Foundation of China (Grant Nos.81672479, 81802483, and 81872054), the National Postdoctoral Program for Innovative Talents (Grant No. BX20180384),and the Liao Ning Revitalization Talents Program (Grant No.XLYC1807255).
Conflict of interest statement
No potential conflicts of interest are disclosed.
Author contributions
Conceived and designed the analysis: Tao Jiang and Anhua Wu.
Collected the data: Zhiliang Wang, Zheng Wang, and Guanzhang Li.
Contributed data or analysis tools: Zheng Zhao, and Chuanbao Zhang.
Performed the analysis: Zhiliang Wang and Wen Cheng.
Wrote the paper: Zhiliang Wang and Wen Cheng.
Availability of data and materials
The expression data were collected from the Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn/index.jsp), The Cancer Genome Atlas (TCGA, (http://cancergenome.nih.gov/)database, and the Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse16011).
 Cancer Biology & Medicine2022年4期
Cancer Biology & Medicine2022年4期
- Cancer Biology & Medicine的其它文章
- N6-methyladenosine (m6A) RNA modification in tumor immunity
- Methods for monitoring cancer cell pyroptosis
- Research progress in hepatitis B virus covalently closed circular DNA
- Effects of menopausal hormone therapy-based on the role of estrogens, progestogens, and their metabolites in proliferation of breast cancer cells
- Breast cancer screening and early diagnosis in Chinese women
- SHP-1 acts as a tumor suppressor by interacting with EGFR and predicts the prognosis of human breast cancer