
N6-methyladenosine (m6A) RNA modification in tumor immunity
2022-02-04 08:20SiyiZhengHuiHanShuibinLin
Cancer Biology & Medicine 2022年4期
Siyi Zheng*, Hui Han*, Shuibin Lin
1Center for Translational Medicine, Precision Medicine Institute, The First Affiliated Hospital, Sun Yat-sen University,Guangzhou 510080, China; 2Department of Otolaryngology, Center for Translational Medicine, Precision Medicine Institute,The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China
ABSTRACT Growing evidence supports that cancer progression is closely associated with the tumor microenvironment and immune evasion.Importantly, recent studies have revealed the crucial roles of epigenetic regulators in shaping the tumor microenvironment and restoring immune recognition. N6-methyladenosine (m6A) modification, the most prevalent epigenetic modification of mammalian mRNAs, has essential functions in regulating the processing and metabolism of its targeted RNAs, and therefore affects various biological processes including tumorigenesis and progression. Recent studies have demonstrated the critical functions and molecular mechanisms underlying abnormal m6A modification in the regulation of tumor immunity. In this review, we summarize recent research progress in the potential roles of m6A modification in tumor immunoregulation, with a special focus on the anti-tumor processes of immune cells and involvement in immune-associated molecules and pathways. Furthermore, we review current knowledge regarding the close correlation between m6A-related risk signatures and the tumor immune microenvironment landscape,and we discuss the prognostic value and therapeutic efficacy of m6A regulators in a variety of cancer types.
KEYWORDS N6-methyladenosine (m6A) modification; immune evasion; tumor microenvironment (TME); tumor immunology; immune cells
Introduction
Epigenetics, defined as heritable and potentially reversible changes in gene expression that occur without alterations in the DNA sequence, has received increasing attention in the past few decades1. Epigenetic drugs, such as DNA methyltransferase inhibitors and histone deacetylase inhibitors,have been used to restore aberrant levels of epigenetic modifications and have been applied in the clinical treatment of a variety of tumors2. Analogously to DNA and histone modifications, RNA modifications have been shown to regulate gene expression at the post-transcriptional level and have become a major research focus in recent years3. More than 100 post-transcriptional modifications have been confirmed to occur in cellular RNAs. Among them, N6-methyladenosine(m6A), first identified in the 1970s, is the most abundant modification in most eukaryotic mRNAs4. The rapid development of RNA immunoprecipitation-sequencing methods has uncovered the transcriptome-wide m6A modification landscape. The deposition of the m6A modification is not random but occurs mainly in RRACH sequences (where R = A or G,and H = A, C, or U) and is concentrated near the stop codon,3´UTR, and long internal exons5. The m6A modification regulates a variety of biological functions, including tissue development6, circadian rhythms7, the DNA damage response8,and sex determination9. Accumulating studies indicate that aberrant m6A methylation levels contribute to tumorigenesis and cancer progression. Therefore, targeting dysregulated m6A regulators, similarly to other epigenetic regulators, may provide an attractive potential strategy for cancer therapy.
The interaction between cancer and the immune system is complicated. During tumor progression, cancer cells can evade host immune surveillance and weaken the immune response through various mechanisms, including decreasing the expression of tumor-associated antigens or promoting the formation of an immune-inflammatory microenvironment. Although cancer immunotherapy has been shown to be effective across a broad range of cancer types, only a subset of patients with cancer show clinical responses, because immune checkpoints can be dysregulated or hijacked as a mechanism of immune resistance. Therefore, revealing novel molecular mechanisms on tumor immune resistance can improve the efficacy of tumor immunotherapy.
In recent years, epigenetic drugs such as inhibitors of DNA methyltransferases and histone deacetylases have been demonstrated to effectively reverse immune suppression. Those epigenetic inhibitors functionviaseveral mechanisms, such as enhancing the expression of tumor-associated antigens, components of the antigen processing and presenting machinery pathways, immune checkpoint inhibitors, chemokines, and other immune-associated genes2.
Recent studies have revealed the correlation between m6A methylation and tumor immunity, thus broadening the understanding of m6A methylation and tumor immune regulation,and improving the efficacy of immune checkpoint blockade therapy and overcoming immune resistance.
In this review, we focus on the interplay among m6A modification, functional maintenance of the immune system, and the landscape of the tumor immune microenvironment, to provide a theoretical foundation for understanding the crosstalk between m6A modification and anti-tumor immunity. In addition, we highlight the promise of combining targeting of m6A regulators and immunotherapy to restore immune recognition and immunogenicity for the effective treatment of cancers.
Composition of the RNA m6A methylase complex
Beyond the epigenetic modification of DNA and histones,RNA modification has gradually become a major research focus. More than 170 RNA chemical modifications have been identified10. m6A is the most prevalent and reversible internal modification in mammalian messenger and noncoding RNAs. Emerging evidence indicates that abnormal m6A levels are strongly associated with tumor initiation and progression in various types of cancers. m6A modification is dynamically regulated by methyltransferases (“writers”)and demethylases (“erasers”), and is recognized by various readers, functions attributed to m6A cover nearly all aspects of mRNA processing, ranging from pre-mRNA splicing,export, translation and stability (Figure 1)11.
Notably, “writers” are composed of a multicomponent methyltransferase complex that deposits m6A modifications.Methyltransferase-like 3 (METTL3), Methyltransferase-like 14(METTL14), and Wilms tumor1-associated protein (WTAP)were initially identified in this complex12,13. Among them,METTL3 is the key methylase that catalyzes methylation, by transferring a methyl from S-adenosine methionine (SAM) to the adenine moiety of the target. METTL14 forms a heterodimer with METTL3 and stabilizes the METTL3-RNA interaction by providing a platform for RNA binding12,13. METTL3 has been found to promote translation of certain oncogenes in human lung cancer14. Interestingly, METTL3 recruits the translation initiation complex to target mRNAs through direct interaction with eIF3b. The METTL3-eIF3h interaction is required for enhanced translation, the formation of densely packed polyribosomes, and oncogenic transformation15.Similarly, WTAP assists in the localization of heterodimers to nuclear plaques, thus facilitating the recruitment of the complex to specific RNA targets16. Apart from these 3 core components, VIRMA/HAKAI/ZC3H13 have also been identified to be part of the entire methyltransferase complex17. Unlike other methyltransferase complexes, METTL16 is a newly discovered m6A “writer”, which binds a range of non-coding RNAs18and interacts with pre-mRNA, thus regulating the splicing process19.
The removal of the m6A modification is performed by demethylases termed m6A “erasers”. The first demethylase, fat mass and obesity-associated protein (FTO), was discovered in mammals in 200820. To date, only 2 m6A demethylation enzymes have been found in eukaryotes. AlkB homologue 5 (ALKBH5), the second identified “eraser”, has been implicated in the reversible removal of methyl groups from the m6A modification specifically. The discovery of “erasers” has brought another dimension to the study of RNA epigenomics.Mechanistically, the unique C-terminal long loop domain in FTO enables it to demethylate the methylated single-stranded DNA or RNA. FTO specifically removes m6A marks from the GGACU and RRACU motifsviaoxidative demethylation activity. Moreover, demethylation mediated by ALKBH5 is critical for RNA splicing21. The dynamic regulation of m6A by methyltransferases and demethylases provides an additional layer of gene expression regulation at the post-transcriptional level.

Figure 1 Overview of RNA N6-methyladenosine (m6A) modification. The m6A modification is modulated by m6A “writers”, “erasers”, and“readers”. m6A “writers” are methylase complexes including METTL3, METTL14, ZC3H13, RBM15, VIRMA, HAKAI, WTAP, and METTL16. “Erasers”are demethylases (FTO, ALKHB5) that remove methyl groups. The m6A-containing RNAs are recognized by “readers”, which are involved in multiple processes of RNA metabolism, such as primary miRNA processing, alternative splicing, mRNA stabilization, translation, and degradation.
Various m6A “readers” specifically recognize the m6A residues and subsequently exert biological functions in RNA metabolism and multiple biological processes. The m6A“readers” are divided into several protein families: YT521-B homology (YTH) domain family, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs), and heterogeneous nuclear ribonucleoproteins (HNRNPs). YTHDF2, the first recognized “reader”, decreases the stability of targeted transcripts and promotes mRNAs degradation by recruiting the CCR4-NOT deadenylase complex22. Subsequently, more YTH domain proteins were discovered, including YTH domain family proteins (YTHDF1 and YTHDF3), and YTH domaincontaining proteins (YTHDC1 and YTHDC2)23-26. Further studies have indicated that YTHDF3 promotes the translation and degradation of m6A transcriptsviadifferent mechanisms,such as by regulating its own mRNA translation and increasing its protein expression27, as well as interacting with ribosomes and participating in the translation initiation and elongation28.In addition, YTHDC2 has been revealed to regulate the stability of m6A-targeted mRNA and recruit the RNA degradation machinery. Knockdown of YTHDC2 has been demonstrated to increase m6A-modified transcripts29. Moreover, YTHDC1 contributes to RNA splicing and export26. By promoting exon inclusion in targeted mRNAs through recruiting pre-mRNA splicing factor SRSF3 (SRp20) while blocking SRSF10 (SRp38)mRNA binding, YTHDC1 regulates mRNA splicing and consequently mediates the transfer of “targeted mRNAs” from the nucleus to the cytoplasm30.
Huang et al.31have provided compelling evidence that, in contrast to the mRNA-decay-promoting function of YTHDF2,IGF2BPs–a distinct family of m6A “readers” that preferentially recognize thousands of m6A-modified mRNA transcripts through the consensus GG(m6A)C sequence– promotes mRNA stability and translation in an m6A-dependent manner, thus globally affecting gene expression output.Moreover, m6A modification changes the secondary structure of RNA, promotes the binding of HNRNPC and HNRNPA2B1 and other nuclear heterogeneous ribosomal protein family readers, and participates in primary microRNA processing and alternative splicing32.
In short, “writers” and “erasers” determine the m6A levels in specific transcripts; these modifications are interpreted by“readers” and have roles in multiple biological functions and diverse processes in RNA fate, including primary miRNA processing, alternative splicing, and mRNA stabilization, translation, and degradation.
m6A modification involved in immune cells
In the past several decades, m6A methylation has been widely demonstrated to play complex roles in the homeostasis of the immune system, such as the recognition of, and response to,invading pathogens, and exogenous or aberrant endogenous RNAs33-35. Several investigations have demonstrated the crosstalk between m6A regulators and their regulation in general biology in immune cells. Indeed, studies increasingly indicate that RNA m6A methylation is associated with tumor-intrinsic oncogenic pathways. In this section, we review the recent progress in understanding of the roles of m6A modification regulation, with a focus on the anti-tumor processes of both innate and acquired immune cells, including macrophages, dendritic cells, lymphocytes, and natural killer cells.
m6A modification in macrophages
Macrophages are crucial mediators of tissue homeostasis that have phagocytic, antigen presentation, and cytokine secretion functions. Under different tissue microenvironments and cytokines, macrophages can polarize into 2 phenotypes: macrophage type 1 (M1) and macrophage type 2 (M2)36,37. In the tumor microenvironment (TME), M2 has pro- tumorigenic properties38, whereas M1 is involved in promoting anti-tumor immunity39. Recent studies have shown that the m6A modification is involved in regulating the polarization of macrophages. STAT1 is the main transcription factor regulating macrophage polarization into M1. METTL3 increases the transcriptional stability and expression of STAT1 mRNA in an m6A-dependent manner, thus promoting the polarization of macrophages40.
Intriguingly, recent research has suggested that METTL3 is required for macrophage-mediated innate immunity.Mechanistically, METTL3 deficiency significantly decreases m6A modification of IRAKM mRNA, thus diminishing its degradation, resulting in higher IRAKM levels, and suppressing macrophage activationviathe TLR4 signaling pathway41.
Similarly, a recent study has revealed a non-cell-intrinsic,tumor-suppressing function of METTL3: ablation of METTL3 in macrophages has been found to contribute to the formation of an immunosuppressive microenvironment, including increased regulatory T cell (Treg) infiltration, and fewer Th1 cells and IFN-γ+CD8+cells, thus facilitating tumor growth and metastasis. Mechanistically, METTL3 depletion impairs the YTHDF1-mediated translation of SPRED2, thereby enhancing the activation of NF-κB and STAT3 through the ERK pathway. Moreover, loss of METTL 3 impairs PD-1 blockade therapeutic efficacy in B16 tumors42. Beyond METTL3 loss,a deficiency in Mettl14 or Ythdf2 in macrophages promotes the accumulation of EBI3 mRNA in an m6A-dependent manner, thereby triggering CD8+T cell dysfunction, preventing CD8+T cell infiltration, and accelerating tumor progression43.
Overall, these studies illustrate that m6A modification in macrophages might serve as a promising target for tumor immunotherapy.
m6A modification in dendritic cells
Dendritic cells (DCs), another antigen-presenting cell type involved in the immune response, link innate and adaptive immunity. DCs interact with T cells in both tissue lymph nodes and TME, thus promoting functional activation44.Dysregulation of DC-mediated immune activation is generally accepted to be closely associated with several pathological conditions. DC vaccines also have been a promising strategy for cancer treatment in recent years. Exploration of the regulatory mechanisms of the m6A epi-transcriptome has increased understanding of m6A-mediated DC function and supported translational exploitation of m6A-based tumor immunotherapy.
Recently, RNA m6A modification involved in DCs has been reported to be critical for anti-tumor immunity effects.Loss of YTHDF1 in DCs enhances the cross-presentation of tumor antigens and the cross-activation of CD8+T cellsin vivo. Mechanistically, YTHDF1 increases the translation of lysosomal protease transcripts with m6A modification. In addition, the therapeutic efficacy of PD-L1 checkpoint blockade immunotherapy is greater inYthdf1–/–mice than wild-type mice, thus indicating that YTHDF1 is a potential therapeutic target in anti-tumor immunotherapy45.
m6A modification in lymphocytes
Two types of lymphocytes are critical in specific immune responses: T lymphocytes (T cells) and B lymphocytes (B cells).Research on the involvement of m6A modification in lymphocyte development and tumorigenesis remains in early stages.However, studies have focused on the crosstalk between RNA m6A methylation and abnormalities in lymphocyte homeostasis. For instance, a study by Li et al.46has demonstrated that deletion of METTL3 in CD4+T cells inhibits activation of the IL-7/STAT5 signaling pathway, which is associated with the differentiation and development of T cells and contributes to imbalanced naïve T cell homeostasis. Moreover, another study has uncovered the effects of m6A methylation in Tregs,thus suggesting that METTL3 deficiency in Tregs contributes to elevated expression of Socs genes targeting the IL-2/STAT5 signaling pathway47. In addition, YTHDF1 deficiency in DCs intensifies the early steps of T cell priming against tumor neoantigens. Furthermore, the anti-tumor response is completely abrogated inYthdf1–/–mice lacking CD8+T cells, thus highlighting the involvement of m6A modification in the regulation of CD8+T cells and tumorigenesis. Similarly, RNA m6A modification has been demonstrated to control the early development of B cells. More recently, a study has indicated that deletion of METTL14 specifically in B cells leads to severe defects in B cell development in mice, with inhibition of interleukin-7-induced pro-B cell proliferation and blocking of the large-to-small pre-B cell transition48. These studies have demonstrated the important regulatory roles of m6A modification in T cells or B cells as well as their involvement on tumor progression.
m6A modification in NK cells
Natural killer (NK) cells, the predominant innate lymphoid cells, are critical mediators of host immunity against virusinfected and cancerous cells49. In general, NK cells release cytotoxic molecules, which induce apoptosis or pyroptosis. In addition, NK cells enhance the functions of other innate and adaptive immune cellsviathe secretion of several cytokines and chemokines50.
A recent report by Ma et al.51has indicated that YTHDF2 positively regulates NK cells’ anti-tumor and antiviral activity.Briefly, YTHDF2 promotes the secretion of perforin, granzyme B, and IFN-γ, and decreases melanoma metastasis. Depletion of YTHDF2 in NK cells significantly impairs NK cell anti-tumor and antiviral immunityin vivo. Moreover, YTHDF2 has been demonstrated to maintain the homeostasis and maturation of NK cells. Subsequently, a related study unveiled a critical role of METTL3-mediated m6A methylation in the homeostasis and anti-tumor immunity of NK cells. In that study,mice with conditional Mettl3 depletion in NK cells showed aggravated tumor progression and shortened survival, accompanied by suppressed infiltration and impaired tumor immunosurveillance functions of NK cells in the TME52. Overall,these observations have elucidated the role of m6A modification in the anti-tumor immunity of NK cells, highlighting new biological roles of m6A modification in tumor immune regulation in NK cells.
m6A modifications involved in tumor immune evasion
Generally, according to the different characteristics of the TME, tumor immune evasion can be divided into 2 types either with or without a T cell inflamed phenotype. The former type comprises infiltrating T cells and an extensive cytokine profile because of the activation of the immune system. This type resists immune attackviaimmune system suppressive pathways. The latter type lacks infiltrating T cells, exhibiting immune escape by immune system evasion53. Tumor cells can create an immune-inflammatory suppression microenvironment to avoid immune surveillance or weaken the immune response. In addition, the TME regulates tumor progression and drug resistance through altering cell-cell and extracellular matrix adhesion and modulating immune responses.
This section focuses on m6A modifications involved in the regulation of immune-associated molecules and tumor immune-associated pathways involved in the formation of the tumor immune environment and tumor immune evasion, as well as drug resistance.
m6A regulators involved in the expression of immune inhibitory molecules
Programmed death-ligand 1 (PD-L1) is a primary immune inhibitory molecule expressed on tumor cells that promotes immune evasion. In the past few years, immune checkpoint blockade therapies blocking programmed cell death protein 1 (PD-1) and PD-L1 have shown tremendous benefit in patients with various types of tumors. The PD-1/PD-L1 blocking response is associated with numerous tumor-intrinsic and tumor immune microenvironment characteristics54,55; as a result, a considerable proportion of patients show no response or develop resistance to PD-1/PD-L1 blockade therapy. Therefore, further understanding of the molecular mechanism of PD-1 regulation may aid in enhancing the therapeutic efficacy of checkpoint blockade.
Compared with untreatedYthdf1−/−mice or anti-PD-L1-treated WT mice,Ythdf1−/−- mice treated with PD-L1 checkpoint blockade immunotherapy have been found to show more complete tumor regression, thus suggesting YTHDF1 as a potential target in anti-tumor immunotherapy. In addition,Wang et al.56have shown that, in colorectal carcinoma and melanoma, depletion of METTL3 and METTL14 enhances the response to anti-PD-1 blockade therapy through stabilizing Stat1 and Irf1 mRNA. These findings have increased awareness of the function of RNA methylation in response to anti-PD-1 treatment, and suggested METTL3/14 and YTHDF1 as potential therapeutic targets in anti-PD-1 checkpoint blockade immunotherapy.
Remarkably, inhibition of FTO in melanoma has been found to suppress tumorigenicity and to increase the m6A levels in PD-1, CXCR1, and SOX10, hence enhancing the decay of these m6A-targeted mRNAs by YTHDF2. More importantly,in vivo, selective blocking of FTO restores the IFN-γ response and increases sensitivity to anti-PD-1 therapy treatment57. Similarly, Tsuruta et al.58have revealed that the RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Depletion of FTO decreases PD-L1 expression in an IFN-γ signaling-independent manner.This result has provided new insights into the regulation of PD-L1 expression by RNA modification. A recent study has also indicated that in intrahepatic cholangiocarcinoma, tumor-intrinsic ALKBH5 inhibits the cytotoxicity of T cells by sustaining tumor cell PD-L1 expression. Moreover, ALKBH5 decreases the infiltration of myeloid-derived suppressor-like cells in the tumor immune microenvironment, and the ALKBH5-PD-L1-regulating axis has been confirmed59. Together, these studies have identified FTO and ALKBH5 are direct targets of PD-1 or PD-L1, and have revealed a new PD-L1 regulatory mechanism involving mRNA epigenetic modification. More importantly,these findings provide a basis for substantially improving the therapeutic efficacy of checkpoint blockade therapy and inhibiting tumor immune evasion.
Beyond PD-L1, CD47 is a key receptor expressed in tumor cells and is involved in immune escape in the TME. CD47 is a transmembrane glycoprotein well known for its “self/do not eat”signal in normal cells. Mechanically, the interaction between CD47 and macrophage signal regulatory protein alpha (SIRPα)inhibits phagocytosis by macrophages. The level of CD47 has also been reported to correlate with invasion and metastasis in various malignancies including leukemia, lymphoma, multiple myeloma, and several solid tumors, such as breast cancer and hepatocellular carcinoma (HCC)60. Most recently, Fan et al.61have revealed the regulation of CD47 expression in HCC.Mechanically, METTL3/IGF2BP1 positively regulate CD47 expression in an m6A-dependent manner, and CD47 mediated EMT has been found to contribute to microwave ablation-induced metastasis in HCC. These findings shed new light on the crosstalk between CD47 and m6A modification.
m6A modification involved in immuneassociated signaling pathways
Apart from the above immune inhibitory molecules, several immune-associated signaling pathways also regulate the tumor immune microenvironment. Here, we summarize the crosstalk between m6A modification and key molecules in immune-associated signaling pathways, and discuss the related potential therapeutic strategies.
HIPPO/YAP signaling pathway
The HIPPO signaling pathway plays a crucial role in priming the appropriate immune responses to viruses, bacteria, and several carcinogenic factors for the maintenance of homeostasis, among which the core regulatory protein Yes-associated protein (YAP) participates in tumor immunoregulation.When YAP is in a low phosphorylation state, it functions as a transcriptional co-activator promoting downstream target gene expression. Activated YAP is beneficial in recruiting tumor-associated macrophages62.
In prostate cancer cells and pancreatic ductal cancer cells,high expression of YAP upregulates the secretion of the CXCL5 chemokine, promotes the recruitment of MDSCs, and protects tumor cells from immune clearance63,64, whereas phosphorylation and inactivation of YAP lead to a decrease in Treg number and activity, thereby promoting anti-tumor effects65.In addition, YTHDF2 orchestrates the epithelial- mesenchymal transition/proliferation dichotomyviaHIPPO/YAP signaling,thus suggesting that YTHDF2 has potential value as a target for pancreatic cancer66. These findings demonstrate that YTHDF2 is involved in tumor immune evasion.
YAP activation is crucial for cancer progression, and m6A modified transcripts of lncRNA have been demonstrated to regulate YAP activation in colorectal cancer (CRC). In CRC tissue, YTHDF3 expression is elevated. YTHDF3 recognizes m6A-containing GAS5 and accelerates its degradation, thus further activating the YAP signaling pathway in CRC67. In addition, Zhang et al.68have uncovered a novel circRNA_104075 related pathway that stimulates YAP in HCC and have provided new evidence that m6A modification in the YAP 3´UTR participates in HCC occurrence and development.More recently, studies have shown that m6A modification regulates the expression of YAP and may have potential clinical importance in the diagnosis and prognosis of several tumor types such as non-small cell lung cancer (NSCLC). In NSCLC,METTL3 increases the stability and the efficiency of translation of YAP by upregulating YAP m6A levels and recruiting YTHDF1/369. Similarly, another study has revealed that, in NSCLC, ALKBH5 modulates YTHDF-mediated YAP expression by removing m6A modifications on YAP, and inhibits YAP activity through the miR107/lats2 axis, thus suppressing tumor growth and metastasis70. Therefore, effective regulation of YAP m6A levels may be a potential therapeutic strategy for NSCLC. Overall, m6A modification has been reported to be involved in tumor progression and metastasis through the HIPPO signaling pathway in several studies.
WNT/β-catenin signaling pathway
Similarly to the HIPPO/YAP signaling pathway, the WNT/βcatenin signaling pathway plays a pivotal role in the maintenance of cellular homeostasis and the functional regulation of several immune cells. It also regulates immunosurveillance mechanisms in the TME under various physiological conditions. Alterations in this pathway are associated with the dysregulation of the anti-tumor immune response. Abnormal activation of the WNT/β-catenin signaling pathway has been observed in a wide range of solid tumors of non-T-cell inflammatory infiltration types, such as bladder cancer71, head and neck squamous cell carcinoma72, epithelial ovarian cancer73,and CRC74, beyond metastatic melanoma. In tumor cells with high expression of β-catenin and activation of the WNT pathway, CCL4 secretion is diminished, and the recruitment of DCs into the TME by CCL4 is blocked, thus posing an obstacle to T cell infiltration into the TME75. Overall, the WNT signaling pathway is essential for tumor progression and survival,even in the presence of the anti-tumor immune response76.
Recently, anomalous expression of m6A regulators has been revealed in cancers with abnormal activation of the WNT signaling pathway. For example, in CRC, YTHDF1 recognizes and promotes the translation of methylated FZD9 and WNT6, thus increasing the expression of β-catenin and abnormal activation of the WNT/β-catenin signaling pathway.Knockout of YTHDF1 significantly inhibits the development of CRC77. Similarly, METTL3 targets CTNNB1 in hepatoblastoma. Mechanistically, methylation of CTNNB1 transcripts by METTL3 increases the expression of β-catenin, which is involved in the regulation of tumor immunity, thereby increasing the proliferation and tumorigenicity of hepatoblastoma cells78. Additionally, in osteosarcoma tissues and cell lines compared with in normal tissue cells, METTL3 is more highly expressed, and it regulates the m6A level of LEF1 and activates the WNT/β-catenin signaling pathway, thus playing a critical role in the progression of osteosarcoma79. Moreover,in HCC, YTHDF1 promotes the translational output of FZD5 mRNA in an m6A-dependent manner and facilitates the progression of HCC, thus acting as an oncogeneviathe WNT signaling pathway80.
Therapeutic and prognostic efficacy of m6A regulators
Currently, cancer treatments often include surgery, chemotherapy, radiotherapy, and immunotherapy, although therapy resistance has been demonstrated to be a consequence of multiple factors such as individual variability in drug sensitivity,tumor location, tissue lineage, tumor aggressiveness, and intracellular molecular alteration81. The major reason for the failure to eliminate tumors is a lack of comprehensive understanding of the molecular mechanism underlying therapeutic resistance.
The PD-1 blocking response is associated with numerous tumor-intrinsic and tumor immune microenvironment characteristics54,55; as a result, a considerable proportion of patients show either no response or drug resistance. Thus, searching for biomarkers predictive of the immunotherapy response and identifying patients who may benefit from treatment might guide more effective immunotherapy strategies and the development of promising therapeutic targets.
Emerging evidence indicates that in multiple cancer types,m6A regulators are strongly associated with acquired therapy resistance, including acquired chemoresistance, radioresistance,and resistance to immunotherapy81. For instance, in melanoma and CRC, ALKBH5 regulates the anti-PD-1 therapy response by modulating lactate levels and suppressing immune cell infiltration in the TME. Mechanistically, ALKBH5 regulates the expression of Mct4/Slc16a3 and the content of lactate in the TME,thus suppressing the composition of tumor-infiltrating Treg cells as well as MDSCs, and finally regulating the response of PD-1 immunotherapy82. In another example, LILRB4 targeted by FTO in AML promotes immune evasion83. In melanoma,inhibition of FTO suppresses tumorigenicity and increases the m6A levels in PD-1, CXCR1, and SOX10, hence enhancing the decay of these m6A-targeted mRNAs by YTHDF2. More importantly, selective blocking of FTO restores the IFN-γ response and sensitizes anti-PD-1 therapy treatmentin vivo57.
Recent studies have consistently demonstrated that the m6A-related risk signature (“m6A score”), defined by multiple m6A regulators that vary among cancer types, may be developed as an independent prognostic factor for acquired therapy resistance against immune checkpoint inhibitors.
Integrating multiple m6A regulators’ modification patterns to evaluate and identify different TME landscapes has revealed that m6A modification is important in shaping different TME landscapes. For instance, in gastric cancer, m6A modification patterns mediated by multiple m6A regulators are highly correlated with several immune-associated biological processes, thus leading to the apparent heterogeneity and complexity of immune cell infiltration characteristics in the immune microenvironment of gastric cancer84. Another study has demonstrated the correlation between m6A modification and the immune landscape in ccRCC and verified that the m6A score is an independent prognostic factor for PD-1 blockade therapy response in patients with advanced ccRCC85.Additionally, after systematic analysis of 21 m6A regulators in adrenocortical carcinoma samples from TCGA and GEO databases, Jin et al.86have identified 3 m6A modification patterns with various clinical outcomes, demonstrating that the m6A clusters are strongly associated with immune infiltration in adrenocortical carcinoma. More recently, Xu et al.87have confirmed a significant difference in m6A regulator levels between patients with high and low lung squamous cell carcinoma (LUSC). Compared with low-risk LUSC patients,high-risk LUSC patients have significantly lower rates of ALKBH5, METTL3, HNRNPC, and KIAA1429, and display more promising responses to anti-PD-L1 immunotherapy.Subsequently, through developing the five m6A regulators as a prognostic risk signature in lung adenocarcinoma, another study has similarly shown that patients with high-risk, rather than low-risk, lung adenocarcinoma have significantly higher PD-L1 levels, a lower proportion of CD8+T cells, and better response to checkpoint blockade therapy, thus indicating that these five m6A regulators are highly correlated with immune infiltration levels and the immune checkpoint blockade response88. Similar studies have been performed in breast cancer and pancreatic ductal adenocarcinoma in addition to the above tumor types. He et al.89have categorized 775 breast cancer patients into 2 subgroups and revealed that the high methylation group showed higher expression of tumor-infiltrating CD8+T cells and activated NK cells, but less PD-L1, PD-L2,TIM3, and CCR4 than patients in the lower RNA methylation group. Moreover, tumors with high m6A scores are characterized by diminished immune infiltration and T cell exhaustion in patients with pancreatic ductal adenocarcinoma90. Briefly,other analyses in head and neck squamous cell carcinoma and esophageal cancer have linked the m6A score with the tumor immune microenvironment landscape, thus confirming that the m6A score is suitable for development as an immune therapeutic target and viable prognostic biomarker91,92.
Together, increasing evidence demonstrates that m6A modification patterns in a variety of cancer types have an indispensable role in overall TME infiltration characteristics and immune evasive phenotypes. A comprehensive evaluation of m6A modification patternsviatheir interactions with multiple m6A regulators in individual patients would not only enhance understanding of the tumor immune landscape but also provide novel insights regarding current immunotherapeutic strategies. Targeting m6A regulators or m6A phenotypeassociated genes to restore the m6A modification level and reverse aberrant TME cell infiltration may provide effective anti-tumor therapeutics for patients with cancer.
Conclusions and perspectives
In recent years, extensive epigenetic studies have gradually increased knowledge regarding the pivotal role of m6A methylation and its underlying molecular mechanisms in cancers.Currently, m6A modifications have been demonstrated to play crucial roles in metabolism, drug resistance, and metastasis in a variety of malignant tumors. Extensive research efforts focusing on m6A regulators have confirmed that targeting dysregulated m6A regulators is an attractive strategy for cancer therapy. In recent years, specific small molecule inhibitors of m6A modification have gradually been developed.
In the past few years, small molecule inhibitors targeting FTO, such as the natural product rhein93and meclofenamic acid94, have been identified and used to regulate aberrant m6A levels. However, more effective and selective inhibitors have been discovered, including FB23, FB23-295, CS1, CS283, and FTO-0496. In addition, one study has identified 2 inhibitors targeting ALKBH5, 2-[(1-hydroxy-2-oxo-2-phenylethyl) sulfanyl]acetic acid and 4-{[(furan-2-yl)methyl]amino}-1,2-diazinane-3,6-dione, through high-throughput virtual screening.The efficacy of these compounds has been validated on the basis of suppressed proliferation observed in 3 leukemia cell lines97. Recently, Yankova et al.98have found that STM2457, an effective and selective inhibitor of METTL3, displays promising anti-leukemia effects. Moreover, STM2457 exerts effects in a PDX model and a primary mouse model. In summary, targeting multiple m6A regulators should contribute to establishing more effective treatments for cancer patients and provide new insights into RNA m6A modification in cancer.
Our review is limited by the insufficient studies providing details of the direct connection between the TME and m6A methylation. The molecular mechanisms underlying how these m6A regulators interact with one another in a variety of cancers, and how they regulate the anti-tumor immune response, must be further explored. First, the establishment of more stable and sensitive sequencing technology will be essential. To date, m6A-sequencing technology cannot reach the single-cell level. However, an RNA methylation omics map at the single-cell level will be needed to uncover the detailed interplay between RNA epigenetics and tumor immunology. In the future, an m6A modification omics map at the single-cell level of immune cells in tumor tissues, adjacent tissues and lymph nodes should enable additional advances.
In summary, m6A methylation is closely associated with immunological suppression and tumor progression(Figure 2). A better understanding of the crosstalk between m6A modification and tumor immunoregulation is important to reveal new pathogenic pathways and develop promising therapeutic targets for cancers.
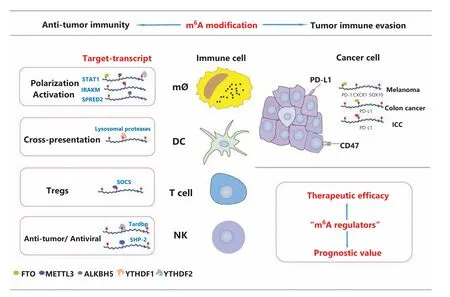
Figure 2 Key regulation of m6A modification in tumor immunity. As shown, the m6A modification is involved in anti-tumor immunity and tumor immune evasion. m6A methylation plays an essential role in the maintenance of immune cell homeostasis and function. Moreover, m6A modification patterns have an indispensable role in overall TME infiltration and the immune evasive phenotype. The close correlation between m6A-related risk signatures and the tumor immune microenvironment landscape supports the prognostic value and therapeutic efficacy of m6A regulators in a variety of cancer types.
Grant support
This research was supported by grants from the National Natural Science Foundation of China (Grant Nos. 81922052,81974435, and 81772999), Natural Science Foundation of Guangdong Province (Grant No. 2019B151502011), and the Guangzhou People’s Livelihood Science and Technology Project (Grant No. 201903010006).
Acknowledgements
We sincerely thank Dr. Demeng Chen for linguistic assistance during the preparation of this manuscript.
Conflict of interest statement
No potential conflicts of interest are disclosed.
Author contributions
Designed and wrote the manuscript: Siyi Zheng, Hui Han.Revised the contents of the manuscript: Shuibin Lin.
 Cancer Biology & Medicine2022年4期
Cancer Biology & Medicine2022年4期
- Cancer Biology & Medicine的其它文章
- Methods for monitoring cancer cell pyroptosis
- Research progress in hepatitis B virus covalently closed circular DNA
- Effects of menopausal hormone therapy-based on the role of estrogens, progestogens, and their metabolites in proliferation of breast cancer cells
- Breast cancer screening and early diagnosis in Chinese women
- SHP-1 acts as a tumor suppressor by interacting with EGFR and predicts the prognosis of human breast cancer
- PGK1-coupled HSP90 stabilizes GSK3β expression to regulate the stemness of breast cancer stem cells