
误诊为结核性脑膜炎自身免疫性胶质纤维酸性蛋白星形细胞病1例
2022-02-03 05:48:14蔡彤彤朱勇冬周厚仕林麒张玉虎
中国神经精神疾病杂志 2022年11期
蔡彤彤 朱勇冬 周厚仕 林麒 张玉虎
自身免疫性胶质纤维酸性蛋白星形细胞病(autoimmune glial fibrillary acidic protein astrocytopathy)是一种新近发现的中枢神经系统自身免疫性疾病,临床上相对罕见、表现多样且无特异性。现报告汕头市中心医院神经内科 2021年5月收治的1例误诊为结核性脑膜炎的GFAP星形细胞病患者,分析其临床表现、影像特征、诊疗情况,以期提高临床工作者对其认识,避免误诊及漏诊。
1 临床资料
患者,男,58岁,因“发热1个月,加重伴乱语3 d”于2021年5月1日入院。患者入院前1个月无明显原因出现发热,多于午后及夜间出现,最高未超过38°C,伴有盗汗、体质量下降,服用抗感染、解热镇痛药物第二天晨起体温可恢复至正常,未引起重视。3 d前出现高热,体温最高达39.5°C,伴有乱语,而来我院急诊,拟“发热、乱语查因”收入我科。自起病以来,患者轻微头痛,无头晕,无恶心、呕吐,无畏寒、寒颤,食欲睡眠差,二便正常,体质量较前下降约3 kg。
入院查体:体温38.6℃,脉搏105次/min,呼吸20次/min,血压120 mmHg /75 mmHg,神志清楚,精神疲乏,定向力正常,记忆力和计算力粗测略有下降,双侧瞳孔等圆等大,直径约2.5 mm,对光反射灵敏,鼻唇沟对称,构音清,咽反射存在,舌居中,四肢肌张力正常,四肢肌力4+级,双侧指鼻试验准确,跟膝胫试验及 Romberg征不配合,四肢腱反射(+),双侧Babinski征(-),脑膜刺激征可疑阳性。
入院诊治:三大常规、生化全套、甲状腺功能、止凝血、传染病八项、肿瘤标志物、风湿三项、免疫五项、抗核抗体谱、降钙素原、PPD、T-SPOT均未见异常,ESR 39 mm/h(正常值0~20 mm/h)。胸部+全腹部CT示:①双肺下叶少许炎症,主动脉硬化;②脂肪肝;③左肾小结石,前列腺钙化。全身PET-CT未见异常代谢征象。24 h视频脑电图,正常范围脑电图。腰椎穿刺,脑脊液压力210 mmH2O(正常值80~180 mmH2O)、常规检查+生化组合示,白细胞0.140×109/L(正常值0~0.008×109/L),淋巴细胞比例99%,氯化物116.1 mmol/L(正常值120~132 mmol/L),葡萄糖 3.07 mmol/L(正常值 2.8~4.5 mmol/L)(对应血糖 6.94 mmol/L),蛋白1.61 g/L(正常值0.15~0.45 g/L),涂片找菌未见异常;脑脊液病原微生物高通量二代测序:阴性。头颅MRI+增强:左侧额叶皮层下、放射冠及双侧侧脑室旁少许腔隙性梗死灶,见图1。
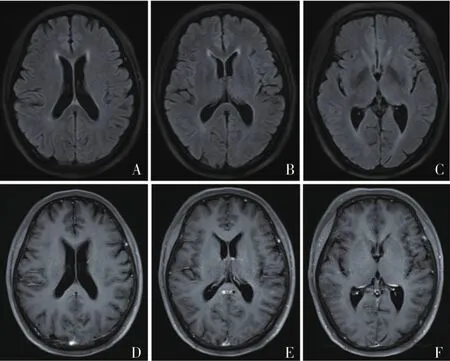
图 1 入院后首次头部MRI+增强 仅可见左侧额叶皮层下、放射冠及双侧侧脑室旁少许腔隙性梗塞灶,在增强上(D~F)无明显强化。
入院后给予阿昔洛韦及头孢曲松抗病毒、抗感染,激素(地塞米松10 mg,每天1次)抗炎,甘露醇脱水。患者仍有发热,体温最高达40°C,伴有畏寒及肢体抖动,意识障碍加重(昏睡),复查腰椎穿刺,脑脊液压力150 mmH2O、常规检查+生化组合示白细胞0.138×109/L,淋巴细胞 97.8%,氯化物 115.3 mmol/L,葡萄糖 1.8 mmol/L(对应血糖5.0 mmol/L),蛋白 0.91 g/L,涂片找菌未见异常,脑脊液病原微生物二代测序阴性,结核分枝杆菌及利福平耐药检测(XPERT MTB/RIF)阴性。考虑患者入院前1个月已存在发热、盗汗、消瘦等结核中毒症状,脑脊液淋巴细胞增多、葡萄糖和氯化物下降、蛋白升高,积极抗感染、抗病毒、抗炎并未奏效,给予经验性抗结核治疗,增加地塞米松用量至20 mg抗炎,第二天患者体温恢复正常,复查头颅MRI+增强:双侧侧脑室、基底节区、丘脑斑片状、条索状异常信号,提示炎症可能,见图2。再次行腰椎穿刺:脑脊液压力、白细胞、葡萄糖和氯化物正常,蛋白1.45 g/L,中枢神经系统脱髓鞘四项中脑脊液抗GFAP抗体阳性(1:100)。

图2 入院后复查头部MRI+增强 可见双侧侧脑室、基底节区、丘脑斑片状、条索状T2- flair高信号(B、C,黑色箭头)在增强上(D-F)可见轻微强化。
最后诊断:自身免疫性GFAP星形胶质细胞病。予甲泼尼龙及免疫球蛋白冲击治疗,患者症状好转出院,激素逐步减量,加用吗替麦考酚酯加强免疫抑制治疗。3个月后复诊,复查头颅MRI增强提示病灶基本消退,脑脊液抗GFAP抗体阳性(1:32)。
2 讨论
胶质纤维酸性蛋白(glial fibrillary acidprotein, GFAP)具有调节突触、维持星形胶质细胞形态稳定、参与血脑屏障形成等生物学功能[1]。2016年首次将GFAP免疫球蛋白G(GFAP immunoglobulin G,GFAP-IgG)作为特异性标记物命名自身免疫性胶质纤维酸性蛋白星形细胞病(autoimmune glial fibrillary acidic protein astrocytopathy)[2]。该病临床相对罕见且表现无特异性,容易误诊及漏诊,常见症状有发热、头痛、意识障碍以及癫痫发作[3]。目前其发病机制尚不完全明确,先前研究提示可能与肿瘤和感染相关。FLANAGAN等[4]的研究队列中有34%的患者检出肿瘤,相较于国外患者,HUANG等[5]纳入的40例患者仅有5例检出肿瘤,本例患者PET-CT检查同样未发现肿瘤。LONG等[6]纳入19例患者,仅有2例患者有单纯疱疹病毒感染,且除单纯疱疹病毒[7]外并未有其他病原微生物被证实与GFAP星形胶质细胞病有关。本例患者两次脑脊液病原微生物高通量测序均阴性,也提示感染并不能完整解释该病的发病机制。因为GFAP是细胞内抗原,GFAP抗体不能在细胞膜和它结合,所以GFAP-IgG是否为致病仍存在争议,目前认为它可能介导了细胞毒性T细胞反应,引起炎症细胞募集,最终使神经组织受到攻击,出现相应神经功能障碍[8-9]。
该病的主要病理改变是血管周围炎症、星形胶质细胞和神经元丢失。血管周围炎症垂直于侧脑室旁分布的特点使我们可以在头颅MRI增强观察到线状和斑片状强化的改变[6]。MRI是该病首选的检查手段,LONG等[6]纳入的19例患者有17例MRI检出异常,其中8例表现为垂直于侧脑室线性增强的特征性改变。FANG等[2]和FLANAGAN等[4]的研究也表明约50%患者在MRI上会出现特征性影像改变,其它非特异的影像表现包括脑室旁和双侧丘脑后部T2-flair高信号、软脑膜及室管膜增强[10]。本例患者复查的头颅MRI可以看到基底节区、丘脑条索状、斑片状T2-flair高信号,但表现并不典型。
GFAP星形胶质细胞病患者脑脊液压力可以正常或者轻度升高,大部分脑脊液常规和生化呈现淋巴细胞增多、蛋白升高的非特异性炎症改变[4]。不同于感染性脑膜炎,因为没有病原微生物的糖酵解消耗,GFAP星形胶质细胞病与其他中枢神经系统免疫性疾病一样,通常不会出现脑脊液葡萄糖和氯化物降低,正是这种类似结核性脑膜炎的脑脊液改变给我们的诊治过程增加了很多困难。虽然本例患者PPD、T-SPOT、XPERT、脑脊液病原微生物高通量测序结果均不支持结核性脑膜炎的诊断,但在抗感染、抗病毒、激素抗炎治疗后脑脊液葡萄糖和氯化物仍进行性下降,病情恶化,加上入院前已有1个月的发热、盗汗、消瘦症状,使得我们不能完全排除结核性脑膜炎的诊断并且经验性地给予了抗结核治疗。YANG等[11]曾报告2例GFAP星形胶质细胞病,2例患者均表现发热、头痛,脑脊液淋巴细胞增多,蛋白升高,葡萄糖下降,但没有像本例患者一样出现氯化物下降。本例患者脑脊液葡萄糖和氯化物下降的原因仍不明确,日本学者[3,12]发现部分GFAP星形胶质细胞病患者脑脊液ADA水平会出现短暂性升高,脑脊液ADA 水平升高通常被认为是结核性脑膜炎的特点,ADA>8 U/L 高度提示结核性脑膜炎[13],但GFAP星形胶质细胞病脑脊液葡萄糖、氯化物下降与ADA升高是否关联目前仍不清楚。
GFAP星形细胞病急性期的治疗主要为激素和丙种球蛋白冲击以及血浆置换,维持阶段的方案主要是激素、免疫抑制剂口服序贯治疗。70%患者对激素反应良好,但有部分患者对治疗反应不佳并且容易复发[14]。对于存在广泛中枢神经系统损伤和抗体高滴度的患者,单纯使用激素或丙种球蛋白冲击通常疗效不佳,对于此类患者,复合血浆交换、蛋白A免疫吸附[15]以及使用免疫抑制可能有临床获益[16],因此,对本例患者,加用吗替麦考酚酯强化免疫抑制治疗。
综上所述,本文报告了1例临床症状、脑脊液改变与结核性脑膜炎非常类似的GFAP星形细胞病,这在临床上非常罕见,极易误诊,这提示在临床上遇到此类患者,应想到GFAP星形细胞病[17-18],尽早完善GFAP-IgG检测并给予有力的免疫抑制治疗,特别是MRI可以看到特征性影像时。
猜你喜欢
传染病信息(2022年2期)2022-07-15 08:55:02
云南化工(2021年11期)2022-01-12 06:06:18
传染病信息(2021年6期)2021-02-12 01:52:58
数学物理学报(2019年3期)2019-07-23 01:15:36
表面工程与再制造(2019年1期)2019-05-11 08:51:52
现代检验医学杂志(2016年2期)2016-11-14 02:38:02
物理化学学报(2015年5期)2015-02-28 17:34:58
湿法冶金(2014年3期)2014-04-08 01:04:51
右江医学(2014年1期)2014-03-22 04:18:28
云南畜牧兽医(2014年2期)2014-02-28 21:25:18
