
Development of an analysis method for determination of pectin in tobacco by solid state 13C CP/MAS NMR
2022-01-26 01:52,,,,,,*
中国科学技术大学学报 2021年8期
, , , , , , *
1. Department of Chemistry,University of Science and Technology of China,Hefei 230026,China;2. Technology Center,China Tobacco Fujian Industrial Co.,Ltd.,Xiamen 361001,China
Abstract:A quantitative analysis method was developed for determination of pectin content in tobacco samples by solid state 13C CP/MAS NMR.A 5.5 mm outer diameter of dimethyl silicone rubber tubing was designed and utilized as a reference of intensity,which was packed into a 5.5 mm inner diameter zirconia NMR rotor to construct an NMR sample tubing.The powder samples filled into the sample tubing were detected to obtain 13C CP/MAS NMR spectra.The peak of C-6 at 171 ppm was processed with spectral deconvolution to eliminate interference from overlapping peaks.The calibration curve was established with the area ratios of assigned C-6 peak to intensity reference peak and the mass of the polygalacturonic acid (PGA).This method was used to determine the pectin content in six different tobacco samples.Relative errors were between -4.94% and 3.84% compared with the results measured by the standard method.The recovery of PGA from spiked tobacco samples was ranged from 94.33% to 102.77%,the RSD (n=5)was less than 2.32%.It demonstrates that the 13C CP/MAS NMR method with a novel intensity reference possesses the properties of speediness,accuracy and simplicity,which is suitable for the quantitative analysis of pectin content in tobacco.
Keywords:pectin;dimethyl silicone rubber tubing;solid state 13C CP/MAS NMR;tobacco
1 Introduction
Pectin designates a family of plant cell wall polysaccharides that is synthesized in the Golgi apparatus before it is secreted into the cell wall as a methylester[1].Pectin is principally made up of four structural units:homogalacturonan (HG),xylogalacturonan,rhamnogalacturonan type I (RGI),and rhamnogalacturonan type II (RGII)[2-4].Homogalacturonan constitutes approximately 65% of the pectin molecule and is composed of an unbranched chain of D-galacturonic acids (GalA)linked by α-1,4 glycosidic bonds[5,6].Therefore,the content of D-galacturonic acid is generally selected as a quantitative indicator of pectin.Pectin is one of the most complex natural heteropolysaccharides,as it can be composed of as many as 17 different monosaccharides containing more than 20 different linkages[7].In food industry,pectin is widely used as a gelling,stabilizing and thickening agent in foods such as jams,jellies,yogurt drinks,fruit milk drinks and ice cream[8,9].In the tobacco industry,pectin is beneficial to the moisture retention of tobacco,accounting for approximately 5% to 13% of the total mass of tobacco leaves.However,the high content of pectin in tobacco will result in incomplete combustion when smoking,and the content of harmful substances such as methane,methanol and carbonyl compounds in tobacco smoke will become higher,which is harmful to the health of smokers[10,11].It can be found that the determination of pectin content in tobacco is of great significance to smokers’safety.Therefore,it is necessary to develop a rapid and accurate method for the quantitative analysis of pectin in tobacco.
At present,the traditional methods for the determination of pectin in tobacco include carbazole colorimetry[12],enzymatic hydrolysis-flow analysis[13]and ion chromatography[14].The carbazole colorimetric method is simple and accurate,but it needs to eliminate the interference of soluble sugars and pigments,and its selectivity is also poor.The enzymatic hydrolysis-flow analysis method is fast,but the conditions for enzymatic hydrolysis are strict.A large number of water-soluble sugars and pigments in tobacco will cause interference and affect the accuracy of the determination results.Ion chromatography owns relatively high selectivity and sensitivity because of the application of electrochemical detector,but the complicated and time-consuming sample pretreatment procedure,such as acidolysis and enzymolysis,cannot be ignored.Because the signal intensity observed is proportional to the number of nuclei that resonates at a given frequency,solid-state NMR spectroscopy can be used not only for structural characterization of materials but also for quantitative analysis[15].In addition,the combination of cross-polarization and magic angle spinning (CP/MAS)can effectively improve the sensitivity and resolution of the13C NMR spectrum[16-18].In recent years,solid state13C CP/MAS NMR spectroscopy has been widely applied to the structural characterization and content detection of cell wall materials such as cellulose[19],lignin[20]and pectin[21,22]in plants.
In order to improve the quantitative accuracy of solid state NMR,to compare different samples,and determine absolute concentrations or purity of samples,a normalization or a reference method has been corroborated to be a feasible solution to eliminate the influence of various factors including the spectrometer,the NMR pulse sequence,the temperature,the probe and the sample itself on the determination results[23].This usually requires the use of a stable intensity reference,providing a peak in the spectrum for which the intensity has an absolute meaning[24].Intensity reference substances for solid-state13C CP/MAS NMR spectroscopy generally include tetrakis-(trimethylsiloxy)-silane(TKS)[25,26],4-(N-methylpyrrolidino)bicycle[3.2.1]-octan-8-one triflate[27],sodium-3-trimethylsilylpropionate (TMSP)[28]etc.However,it is difficult to uniformly mix these intensity reference substances and sample powders during sample preparation,which will cause the signal of intensity reference to be uneven and unstable.In fact,as a reference of signal intensity,it only needs to ensure that the intensity reference substance can be fixed in the rotor and generate stable signals.Hall and Wooten[29]encapsulated the reference substance in a cylindrical cell machined from an aluminium nitride ceramic rod to precisely fit inside the NMR rotor.Keeler and Maciel[30]selected silicone rubber cut into small pieces as the intensity reference material and encapsulated it in a glass capillary.Although these methods do not need to consider the problem of uniform mixing of the strength reference material and the sample,the glass capillary tubing and ceramic cylindrical cell containing the intensity reference materials will be liable to the position deviation during the high-speed spinning in the NMR rotor,which will result in large measurement errors.In short,the previous intensity reference materials are inevitable to have some problems including the difficulty of loading and reusing,or the position deviation during the high-speed spinning.
In this work,the dimethyl silicone rubber tubing was selected as the intensity reference.The outer diameter of the silicone tubing was designed to perfectly match the NMR zirconia rotor so as to guarantee that the intensity reference material did not deviate from its position at a high-speed spinning in the NMR rotor.The spectral deconvolution technique was utilized to eliminate the overlapping peak interference for the target peaks in the NMR spectrum.The pectin extraction was also simpler and faster because of eliminating the interference of soluble sugars,pigments and other impurities on the quantitative results.For the tested samples,the results showed high accuracy in the determination of their pectin contents when compared with standard methods.The13C CP/MAS NMR method with a novel intensity reference was successfully developed for the quantitative analysis of pectin content in tobacco.
2 Experimental
2.1 Materials and chemicals
The tobacco samples with different species in the experiment were obtained from Zimbabwe,Anhui Province of China,Yunnan Province of China,and Guizhou Province of China.Dimethyl silicone rubber tubing (166 mg)was customized by Jingming Silicone Products Co.,Ltd.(Lianyungang City,China).Standard samples of polygalacturonic acid (PGA)(≥ 85%)and 1-naphthol (≥ 99%)were purchased from Aladdin Company (Shanghai,China).Carbazole,sulfuric acid,absolute ethanol,sodium hydroxide,and acetone were all of analytical grades and were purchased from Sinopharm Chemical Reagent Co.,Ltd.(Shanghai,China).Distilled water was used throughout the experiment.
2.2 Extraction of pectin in tobacco samples
The extraction of pectin in tobacco samples were performed by following the method described by Zhu et al[22].10 g of oven-dried tobacco powder was processed by the ultrasound with 100 mL distilled water for 30 min so as to simply remove some soluble sugars and pigments from the tobacco sample.Then,the pH of residue was adjusted to 2.0 with H2SO4(pH 0.5)and maintained for 1.5 h at 85 ℃.The extract was filtered and adjusted pH to 3.5 with 1 mol·L-1NaOH.Next,the ethanol solution (1∶1,v/v)was added to the extract to precipitate pectin.The pectin precipitation was washed with absolute ethanol.Finally,it was kept in a vacuum drying box at 40 ℃ to a constant weight and was collected by being ground into powder,which can pass through a 40-mesh sieve.The powder was stored frozen at -18 ℃ until use.
2.3 Solid-state 13C CP/MAS NMR spectroscopy
High-resolution13C CP/MAS NMR spectra of all samples were obtained by a Bruker AVANCE ΙΙΙ 400 MHz spectrometer(9.4 T)operating at 100.63 MHz for13C and 400.15 MHz for1H,equipped with a 7 mm H/X MAS probe.Each sample was weighed to approximately 170 mg±1 mg and recorded,and then mixed with NaCl powder to fill in cylindrical zirconium dioxide rotors of 7 mm outer diameter with dimethyl silicone rubber tubing inserts as an intensity reference and spun at 4 kHz.The same dimethyl silicone rubber tubing was utilized in the whole experiment.In the cross-polarization process,the radio frequency field strengths were 56.8 kHz for1H and 62.5 kHz for13C.The 90opulse lengths for13C and1H were 4μs and 4.4μs,respectively.All FIDs were subjected to an exponential multiplication function with a line broadening a value of 100 Hz prior to Fourier transform.The cptoss pulse sequence was applied to suppress the spinning sideband.For acquisition of13C CP/MAS NNR spectra,acquisition time of 25 ms,contact time of 2 ms,the optimum recycle delay of 2 s,number of scans 2048,and sweep width of 30 kHz were used.All spectra were referenced to the carbonyl peak of glycine at 175.73 ppm.The NMR spectra of the samples were processed by MestReNova 6.1.1 and PeakFit 4.12 software together.
2.4 Spectral deconvolution
2.5 Quantification of 13C CP/MAS NMR method
After the C-6 peak in the13C CP/MAS NMR spectrum of the sample was performed by deconvolution,the target peaks and overlapping peaks were separated.The area ratio between the assigned C-6 peak and intensity reference peak was calculated,and the polygalacturonic acid (PGA)content of the corresponding samples was obtained according to the established calibration curve equation.Then,the pectin content of tobacco samples can be calculated by the following calculation formula.
(1)
WhereY(%)is the pectin content of tobacco samples;M0(g)is the mass of tobacco samples weighed;M1(g)is the mass of the extracted pectin sample;M2(mg)is the mass of pectin sample loaded in NMR rotor;m(mg)is the mass of PGA calculated from the calibration curve.S(%)is the moisture content;f(%)is the purity of the standard sample of PGA.
2.6 Spectrophotometry
The content of pectin in tobacco was determined by a standard method published by Ministry of Agriculture of the People’s Republic of China (NY/T 2016-2011)[31].5 g of tobacco sample in 50 mL of 75 ℃ absolute ethanol was stirred and heated in a water bath at 85 ℃ for 10 min to extract soluble sugars and pigments.After cooling and filtering,the residue was repeatedly extracted with 67% ethanol in a water bath at 85 ℃for 10 min until soluble sugar was completely removed.Then,the residue was heated and extracted with H2SO4(pH 0.5)in a water bath at 85 ℃ for 1 h.After cooling,the pectin extract was transferred to a 100 mL volumetric flask.Next,the pectin extract was diluted (1∶50)by distilled water,and then 1 mL of the diluent and 0.25 mL of carbazole ethanol solution were added into a test tube.Shake the test tube continuously and quickly add 5.0 mL of sulfuric acid to the test tube.After that,immediately put the test tube in an 85 ℃ water bath.20 min later,the test tube was cooled rapidly with cold water.Finally,the absorbance was measured at 525 nm by spectrophotometry within 1.5 h.
3 Results and discussion
3.1 Design of dimethyl silicone rubber tubing
In order to resolve the inherent problems of previous intensity reference materials,such as the difficulty of sample mixing,loading and reusing,or the position deviation during the high-speed spinning,a dimethyl silicone rubber tubing was ultimately selected as the intensity reference based on the idea of silicone rubber as an intensity reference proposed by Keeler et al[30].According to the requirements of this experiment,the outer diameter of the dimethyl silicone rubber tubing (0.25 mm of thickness)was designed to be 5.5 mm so that it can fit perfectly with the NMR rotor with an inner diameter of 5.5 mm.The structural design drawing and molecular structure of the dimethyl silicone rubber tubing were shown in Figure 1.

Figure 1.Structural desig drawing and molecular structure of the dimethyl silicone rubber tubing.
The properties required of a suitable intensity reference in NMR are as follows :①small line width;②suitable chemical shift in a vacant region of the spectrum for avoiding peak overlaps;③favourable chemical inertness and stability;④convenient separation from the analyte after the NMR analyses;⑤relaxing behavior that permits accurate absolute intensity measurements under the conditions of the NMR technique of interest[24].In this study,the dimethyl silicone rubber tubing met the above the conditions.The chemical shift of13C of the methyl group in the dimethyl silicone rubber tubing was located at 0 ppm,which was far from the chemical shift of most13C in pectin so that the peak of the intensity reference avoided the interference of more overlapping peaks from pectin samples.Furthermore,the line width of its signal peak was also small,and the chemical shift of the dimethyl silicone rubber tubing can always be maintained at 0 ppm without position shift.The sample powder was tightly packed in the centre of the dimethyl silicone rubber tubing and rotors,which will not be loosened or spilled during the high-speed spinning.Compared with the powder reference material,the dimethyl silicone rubber tubing can be handily loaded and reused.Compared with the glass capillary,the dimethyl silicone rubber tubing improved the stability of NMR spectra and the accuracy of the NMR method.
3.2 13C CP/MAS NMR spectrum of pectin
Figure 2 (a,b)showed13C CP/MAS NMR spectra of standard samples of PGA and pectin in tobacco samples after baseline correction and smoothing by MestReNova 6.1.1 software,respectively.As shown in Figure 2,the signal intensities of C-1,C-2,3,5,C-4,C-6 and intensity reference were strong.The resonance signal between 160 ppm and 180 ppm in the spectrum was assigned to the C-6 carbon of galacturonic acid units that were present as carboxylic acid (-COOH),methyl ester (-COOCH3),or carboxylate anion (-COO-).The absorption peak positions of the carboxyl resonance of the protonated (-COOH),esterified forms (-COOCH3)and the ionized form (-COO-)in galacturonic acid were ~171 ppm,~174 ppm and ~176 ppm,respectively[22,32,33].The resonance signals at 101 ppm and 79 ppm were mainly C-1 and C-4 carbon of the glycosidic bonds.The resonance signals of 67 ppm~72 ppm were C-2,3,5 carbon of the pyranoid ring.The intense resonance at 53 ppm in Figure 2 (b)represented methyl carbons of the methyl ester (COOCH3).Signals at 18 ppm in Figure 2 (a)and Figure 2 (b)probably belonged to methyl carbons of rhamnose[34,35].The resonance signal at 0 ppm originated from -CH3of the dimethyl silicone rubber tubing.From the perspective of peak signals and chemical shifts,the peak pattern of the NMR spectrum of pectin in tobacco was basically the same as that of the standard PGA sample.The chemical shifts of the C-6 peak (at ~171 ppm)and the peak of the intensity reference (at ~0 ppm)in the NMR spectra from pectin in tobacco (Figure 2 (b))were very close to the standard sample of PGA (Figure 2 (a)).These results indicated that pectin in tobacco can be characterized by the solid-state13C NMR spectroscopy.Furthermore,the peaks of C-6 and intensity reference in the NMR spectra from tobacco samples were stable,with low interference and strong signals,which were suitable for the quantitative analysis of pectin.

Figure 2.(a)NMR spectra of a standard sample of PGA processed by MestReNova software with Baseline Correctio and Smoothing functions;(b)NMR spectra of pecti i a tobacco sample processed by MestReNova software with Baseline Correctio and Smoothing functions.
The broadening of the C-6 peak was resulted from the superposition of several individual resonances that showed only minor differences in their chemical shifts.Overlapping peaks could be attributed to impurities remained in the pectin extraction process,instrument noise and other factors occupied a non-negligible proportion in the C-6 region,and must be considered because of having the adverse effect on the accuracy of quantitative results of pectin.In the high magnetic field,it is a very direct and effective way to accurately resolve and assign the carbonyl region of the pectin sample by increasing MAS spinning speed or combining with two-dimensional NMR technique[36,37].Unfortunately,these techniques are limited by the conditions of NMR spectrometer so as to be difficult to implement.Generally,the spectral deconvolution can separate the target peaks from the overlapping peaks,then integrate assigned region to increase the reliability of NMR spectral integration[38].Consequently,the deconvolution technique will be an efficient way to resolve the interference of overlapping peaks in the C-6 region.The spectrum optimized by MestReNova 6.1.1 software was imported into PeakFit 4.12 software,and the C-6 peak was performed by selecting method III-Deconvolution and Lorentz Amp/Area peak type to curve fitting.Finally,the respective peaks of target peak and interference peak will be obtained(Figure 3).The target peaks from the carboxyl resonance signals of protonated (-COOH),esterified (-COOCH3)and ionized (-COO-)in galacturonic acid were selected and processed by peak fitting,as shown in Figure 3.Since the fitting peak of the three target peaks represented practical C-6 peak,and it was assigned to be the quantitative peak of pectin.As shown in Figure 4,the intensity reference peak was also processed by spectral deconvolution.On the contrary,the intensity reference peak was not interfered by overlapping peaks.Consequently,the original peak can be used as the quantitative peak of the intensity reference.

Figure 3.Fitting curve of the C-6 peak processed by PeakFit software spectral deconvolutio with Lorentzia profile function.

Figure 4.Fitting curve of the intensity reference peak processed by PeakFit software spectral deconvolutio with Gaussia profile function.

Is there anything I can do to win an immortal soul?” “No,” said the old woman, “unless a man were to love you so much that you were more to him than his father or mother; and if all his thoughts and all his love were fixed70 upon you,23 and the priest placed his right hand in yours, and he promised to be true to you here and hereafter, then his soul would glide into your body and you would obtain a share in the future happiness of mankind
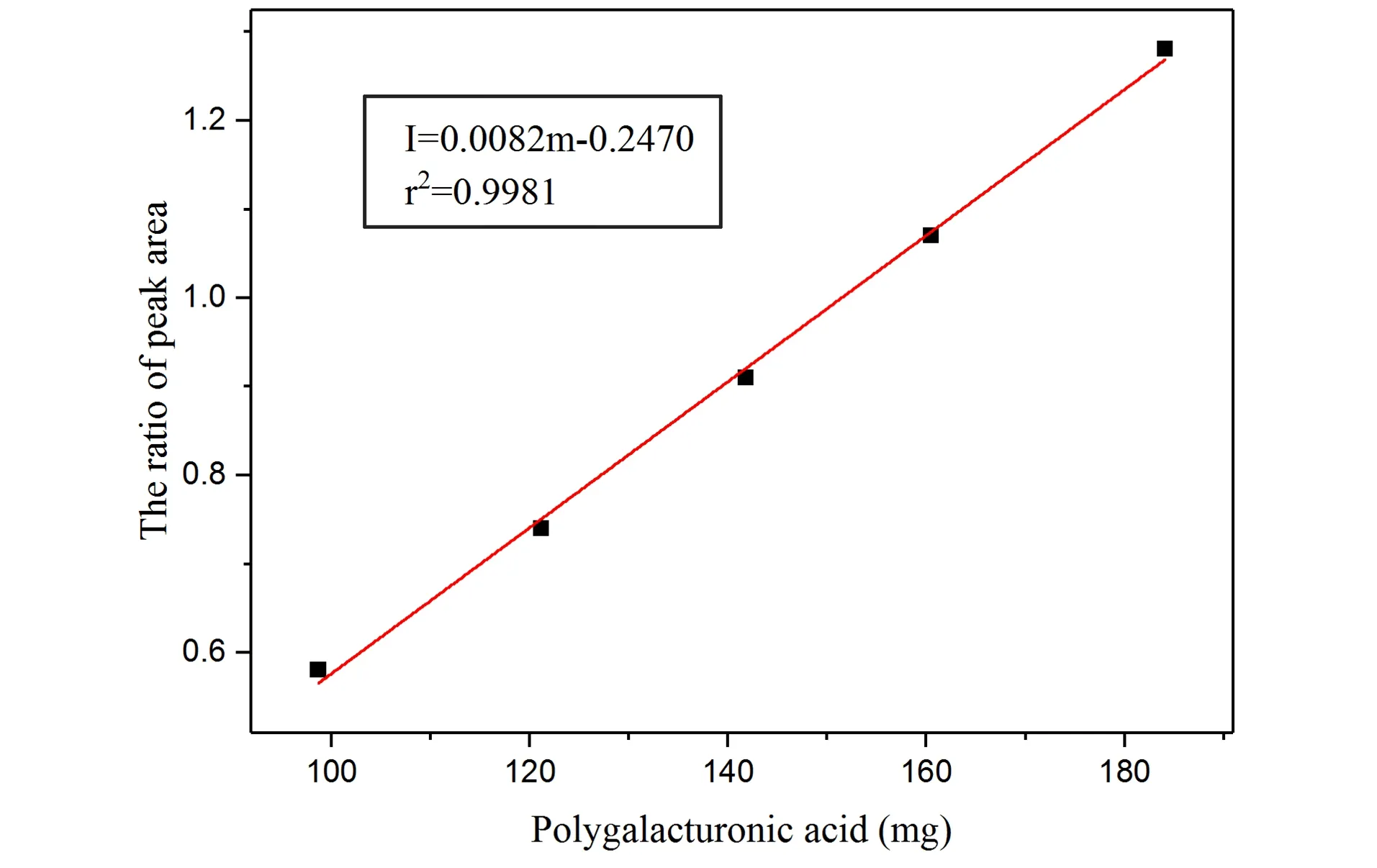
Figure 5.Calibratio curve for the determinatio of PGA content i sample by 13C CP/MAS NMR method with a novel intensity reference.
The area of each peak generally indicates the relative amount of the corresponding functional group in the quantitative analysis by13C CP/MAS NMR spectroscopy[44].Due to the fluctuation of the instrument conditions and the inhomogeneity of the sample,the signal-to-noise ratio of the same batch of samples will vary greatly even if the instrument parameter settings are the same[45].The different degrees of signal distortion will result in the variation of the13C CP/MAS NMR spectrum.However,if a material can provide a stable intensity reference at a fixed chemical shift in a13C CP/MAS NMR experiment,the peak area in the spectrum will be proportional to the number of spins at the respective chemical site and phase and their relative ratios remain unchanged,which is beneficial to enable the quantitative analysis[46].Previous studies have reported that many researchers have successfully carried out the quantification for lignin in wheat straws[28],cellulose in tobacco[29]and hexamethylbenzene[47]by selecting a material as an intensity reference to establish a calibration curve which was based on the mass of standard samples and the signal ratios of standard samples to intensity references.
3.3 The availability evaluation of the target 13C CP/MAS NMR method
To determine PGA content of pectin samples,five standard samples of PGA weighing 98.7 mg,121.2 mg,141.8 mg,160.5 mg and 184.1 mg were evenly mixed with NaCl powder and filled into the designed sample tubing,and the corresponding calibration curve can be established on the basis of their13C CP/MAS NMR spectrums.After C-6 peak in the NMR spectrum was processed by spectral deconvolution to eliminate overlapping peaks,a calibration curve was established according to the area ratio of assigned C-6 peak to intensity reference peak and the mass of the PGA standard sample,as shown in Figure 5.The corresponding calibration equation wasI=0.0082m-0.2470,and the correlation coefficient wasr2=0.9981.The limits of detection (LODs)and limits of quantification (LOQs)were 1.81 mg·g-1and 6.04 mg·g-1,respectively,which were calculated with signal-to-noise ratios of 3 and 10.
To evaluate the accuracy of13C CP/MAS NMR method with a novel intensity reference for the quantitative analysis of pectin in tobacco,50.6 mg,80.6 mg and 120.4 mg of standard sample of PGA were added to three tobacco samples (10 g)for recovery experiments,and the results were shown in Table 1.It can be seen that the recovery of this method ranged from 94.33%to 102.77%,the average recovery rate reached 98.11%,and the RSD (n=5)was less than 2.32%.According to the results of the data analysis,it was found that the target13C CP/MAS NMR method exhibited good precision and accuracy,so it was an accurate method for the quantitative analysis of pectin in tobacco.
3.4 Sample analysis of 13C CP/MAS NMR method
To investigate the reliability of the proposed method for the determination of pectin in tobacco,the developed13C CP/MAS NMR method with a novel intensity reference was performed to determine pectin content in six different tobacco samples.Meanwhile,the results were compared with those determined by spectrophotometry and13C CP/MAS NMR method without intensity reference.Spectrophotometry issued by Ministry of Agriculture of the People’s Republic of China[31]for the determination of the pectin content in tobacco was used as a reference standard in the experiment.The results were shown in Table 2.The comparison of the average pectin content determined by the NMR method with intensity references and spectrophotometry showed that the relative errors of determination results from these two methods were between -4.94% and 3.84%.The RSD (n=3)of NMR method with intensity reference was less than 2.65%.By contrast,comparing the determination results of NMR method without intensity reference and spectrophotometry,it was found that the relative error of only sample 4 was within the range of ±5%,so there were obvious measurement differences between the two methods.Besides,the RSD (n=3)of NMR method without intensity reference was less than 4.28%.Therefore,the accuracy of this research method was good compared with spectrophotometry.Compared with the NMR method without intensity references,this research method had the advantages of higher accuracy and better precision.
Due to the complexity of tobacco sample components,sugars,pigments and other unknown impurities remained in the pectin extraction procedure will be inevitably carried into the pectin sample.These sugars will react with chromogenic reagent and affect the determination results of spectrophotometry[48].Thus,the complete removal of soluble sugars is indispensable for traditional spectrophotometry,and it is a very time-consuming and laborious process.However,the carbon signals of these sugars (such as glucose,fructose and so on)are in the range of 80-110 ppm in the13C CP/MAS NMR spectrum[49,50],so that they do not interfere with the C-6 (171 ppm)peak of pectin.In addition,the spectral deconvolution technique can separate the target peak from the overlapping peaks to overcome the interference of other remained impurities (such as some proteins and organic acids)on the C-6 peak.Therefore,the pectin extraction in this work is simpler and faster than the traditional spectrophotometry for overcoming the various interference factors.

Table 1.The recoveries of three groups of spiked tobacco samples.
Table 2.Detectio content of pecti i tobacco by NMR method with intensity reference,NMR method without intensity reference and spectrophotometry(n=3).

sampleNMR method with intensity referenceAverage pectin±SD(%)RSD(%)Relative error(%)NMR method without intensity referenceAverage pectin±SD(%)RSD(%)Relative error(%)SpectrophotometryAverage pectin±SD(%)RSD(%)16.11±0.121.902.005.46±0.234.12-8.855.99±0.030.5025.41±0.091.713.844.67±0.163.45-10.365.21±0.030.5836.00±0.091.552.746.17±0.223.535.655.84±0.020.3445.40±0.050.83-0.745.19±0.071.26-4.605.44±0.020.3756.04±0.162.65-3.215.47±0.204.28-12.346.24±0.081.2865.00±0.072.21-4.944.93±0.142.77-6.275.26±0.091.71
4 Conclusions
The solid-state13C CP/MAS NMR combined with a novel intensity reference and deconvolution techniques as an innovative method was developed and applied for quantification of pectin in tobacco.The outer diameter of the dimethyl silicone rubber tubing was designed to perfectly match the zirconia rotor so that the dimethyl silicone rubber tubing,namely intensity reference did not appear position shift at a high-speed spinning in the NMR rotor.The spectral deconvolution technology was employed to eliminate interference from overlapping peaks.These approaches greatly improved the accuracy of the13C CP/MAS NMR for the quantitative analysis of pectinin tobacco.For the detection of pectin content in tobacco samples,the results of the target NMR method and standard method were consistent.In addition,the pectin extraction was simple and fast.Therefore,the developed13C CP/MAS NMR method with a novel intensity reference in this study is a fast and accurate quantitative analysis method and is suitable for the determination of pectin content in tobacco.
Acknowledgments
This work was supported by the Science and Technology Project of Fujian China Tobacco Industry Co.,Ltd.(FJZYHZJH2020001).
Conflictofinterest
The authors declare no conflict of interest.
Authorinformation
NIUFanchaois currently a graduate student under the tutelage of Associate Professor YANG Jun at University of Science and Technology of China.His research focuses on the development of new methods for the quantitative determination of biomacromolecules in complex biological samples.
YANGJun(corresponding author)is Associate Professor in the Department of Chemistry of University of Science and Technology of China.His research interests include qualitative and quantitative research and application of biological cell wall macromolecules,the research of reducing tar and harmful components of tobacco.

- 中国科学技术大学学报的其它文章
- 内皮功能失调与泛血管疾病
- LncRNA expression profiles of Schizosaccharomyces pombe in DNA damage inducing environments
- Highly perfluorocarbon loading efficiency of polymer biomimetic nanoparticle encapsulated by erythrocyte membrane to improve tumor phototherapy
- A new standard quadratic optimization approach to beam angle optimization for fixed-field intensity modulated radiation therapy
- Influence of aliovalent doping on the structure and property of Li2MnCl4chloride solid electrolyte
- Effects of specific amino acids on the metabolism of Drosophila melanogaster