
四川山鹧鸪的转录组组装和注释
2022-01-26 07:44:00刘乙何乐为兰月周闯陈本平岳碧松孟杨四川大学生命科学学院生物资源与生态环境教育部重点实验室成都60065四川老君山国家级自然保护区管理局四川屏山645350四川大学生命科学学院四川省濒危动物保护生物学重点实验室成都60065
四川动物 2022年1期
刘乙, 何乐为, 兰月, 周闯, 陈本平, 岳碧松, 3, 孟杨, 3*(.四川大学生命科学学院,生物资源与生态环境教育部重点实验室,成都60065; . 四川老君山国家级自然保护区管理局,四川屏山645350; 3. 四川大学生命科学学院,四川省濒危动物保护生物学重点实验室,成都60065)
四川山鹧鸪隶属鸡形目Galliformes雉科Phasianidae,是中国特有的国家一级重点保护野生动物。由于四川山鹧鸪的分布区域狭窄,种群数量稀少,且长期受人类开发活动造成的栖息地丧失和片段化等因素的影响,其野生种群生存受到较为严重的威胁,被世界自然保护联盟(IUCN)列为濒危(EN)物种(BirdLife International,2016)。此外,IUCN与国际鸟盟、世界雉类协会将四川山鹧鸪纳入鹑类保护组行动计划(IUCN,2014)。四川山鹧鸪仅在中国的四川省和云南省被发现,分布于大相岭山系的南缘、小相岭山系的东缘、凉山山脉的东北部、乌蒙山系的西部。整个区域由10个栖息地斑块和36个潜在栖息地斑块组成,总面积约5 869 km(戴波等,2014)。
分子生态学方面,本课题组研究了四川山鹧鸪线粒体基因组、全基因组和该物种的系统发育(He.,2009;Zhou.,2019),分析了基因组逆转录子和内源性逆转录病毒(Cui.,2016;郑帅等,2019)。系统发育关系研究方面,大部分基于线粒体基因组构建的系统发育树都认同山鹧鸪属是位于雉科的基部位置,是比较原始的类群,山鹧鸪属的单系性得到了支持(Shen.,2009;Kan.,2010)。Chen等(2020)根据山鹧鸪属14个物种的超保守元素、外显子和线粒体基因组构建的系统发育树显示,山鹧鸪属是从非洲迁徙到东南亚定殖的。根据化石和地理模型估算,山鹧鸪属的祖先在中新世早期到达印度支那,但是直到1 000万年前全球冷却加剧时才开始发散为 2个主要分支——“中国分支”和“东南亚分支”。四川山鹧鸪属于“中国分支”的“横断山脉分支”,与环颈山鹧鸪的亲缘关系最近,分化于330万年前。
随着测序技术的发展和成本的下降,单组学和多组学结合的分析将是今后一段时间内的研究重点。本研究对1只成年雄性四川山鹧鸪个体的心脏、肝脏和肾脏进行了转录组测序、组装和注释,为进一步挖掘四川山鹧鸪的功能基因提供基础数据,并为研究该濒危物种提供遗传数据。
1 材料与方法
1.1 组织采样和测序
在四川老君山国家级自然保护区发现受伤的雄性四川山鹧鸪1只,因救助无效死亡。收集心脏、肝脏和肾脏组织,液氮研磨。将组织粉末加入TRIzol试剂(Invitrogen,Carlsbad,CA,USA)提取总RNA。对提取的RNA进行2%琼脂糖凝胶电泳以评估RNA的降解和污染。使用NanoPhotometer分光光度计(Implen,Los Angeles,USA)和带有Qubit 2.0荧光计的Qubit RNA分析试剂盒(Life Technologies,Carlsbad,USA)检查总RNA的纯度和浓度。另外,使用Bioanalyzer 2100上的RNA Nano 6000分析试剂盒(Agilent Technologies,Santa Clara,USA)评估RNA完整性。使用Epicentre Ribo-zero去除rRNA试剂盒(Epicentre,Madison,USA)去除rRNA后,使用Illumina的NEBNext RNA文库制备试剂盒(NEB,Ipswich,USA)构建3个组织的cDNA文库,并在北京诺禾致源公司的Illumina Novaseq 6000平台上以150 bp的配对末端测序长度进行测序。转录组数据以登录号PRJNA638287保存在Gene Expression Omnibus(GEO)数据库中(https://www.ncbi.nlm.nih.gov/bioobject/PRJNA638287)。
1.2 质量控制和转录组组装
使用NGS QC Toolkit v2.3.3(Patel & Jain,2012)和HISAT2 v2.1.0(Kim.,2015)对测序获得的原始序列进行数据质量控制,过滤低质量和带接头的数据。使用MultiQC(Ewels.,2016)评估数据质量。使用Trinity v2.1.1(Grabherr.,2011)组装得到的非冗余干净序列。统计每个基因和转录本的原始读取计数。并使用BUSCO v 5.1.2 (Simão.,2015)评估组装质量。
1.3 转录组注释
转录组注释分为结构注释和功能注释2个部分。使用TransDecoder-v5.5.0(Grabherr.,2011)预测unigenes的蛋白编码区,每个开放阅读框(open reading frame,ORF)的长度最少为100个氨基酸。注释功能基因时,通过BLAST搜索NCBI Non-Redundant Protein Sequences(NR)数据库、Gene Ontology(GO)数据库、KyotoEncyclopedia of Genes and Genome(KEGG)数据库、Swiss数据库和Cluster of Orthologous Group(COG)数据库进行比对,将阈值(E-value)设定为1×10。
2 结果
2.1 测序结果
心脏、肝脏和肾脏的原始序列过滤后分别产生了5.70 G、4.60 G和5.16 G数据(表1)。3个文库的GC含量在48%左右。MultiQC序列质量报告显示,每条序列各位置碱基的测序质量均位于绿色区间,具有平均质量分数的序列数量也是位于绿色区间,说明数据质量很好。每条序列的重复水平均在20%以下的可接受水平。3个文库中没有检测到过表达的序列(1条序列占比超过总数据的1%视为过表达),说明样品未被污染。每条序列各位置的N碱基(无法识别的碱基记为N)比例可忽略不计,除了心脏文库在97位碱基处有0.42%未识别的碱基。接头序列的含量非常低,说明在对原始数据质控时,接头序列被彻底清除。
286 661条转录本经过Trinity组装并去掉冗余后共得到234 488个基因(表2)。使用BUSCO评估组装的完整性为97.6%,在255个核心基因中检测到249个完整的基因(122个单拷贝基因和 127个多拷贝基因),另外6个基因包括4个片段化基因和2个未检出基因。
2.2 基因结构注释
选取每个基因中最长的转录本作为代表序列(unigene),对unigenes的结构分析统计发现,81.29%不包含ORF,16.43%的包含1个ORF,而2.28%的包含2个及以上ORFs。
2.3 基因功能注释
将组装好的基因在主要的5个数据库中进行注释,总共有70 737个基因获得注释结果,占30.17%,其中NR数据库的注释结果最多,COG数据库的注释结果最少(表3)。5个数据库共同注释到的基因6 998个,NR数据库中单独被注释到的基因最多,为23 773个(图1)。

表1 四川山鹧鸪RNA测序数据结果统计Table 1 Statistics of RNA sequencing data results of Arborophila rufipectus
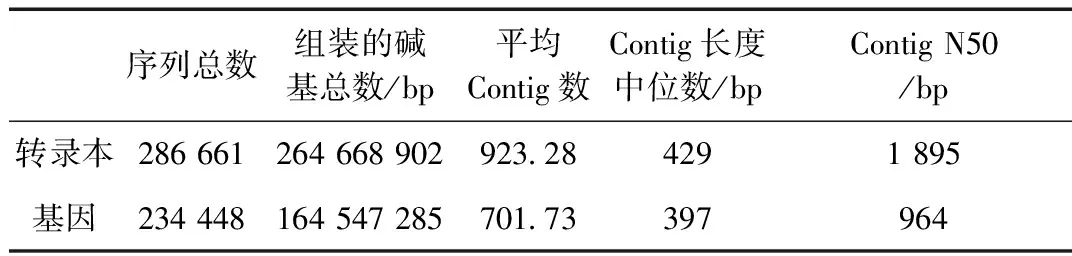
表2 转录本和基因序列信息统计表Table 2 Statistics of transcripts and genes sequences
注: Contig N50: 将所有的Contigs按照从长到短进行排序, 并将Contig按照这个顺序依次相加, 当相加的长度达到Contig总长度的一半时, 最后一个加上的Contig长度即为Contig N50
Notes: Contig N50: sort all Contigs from the longest to the shortest, and add Contigs in this order, when the added length reaches half of the total length of Contig, the last added Contig length is Contig N50
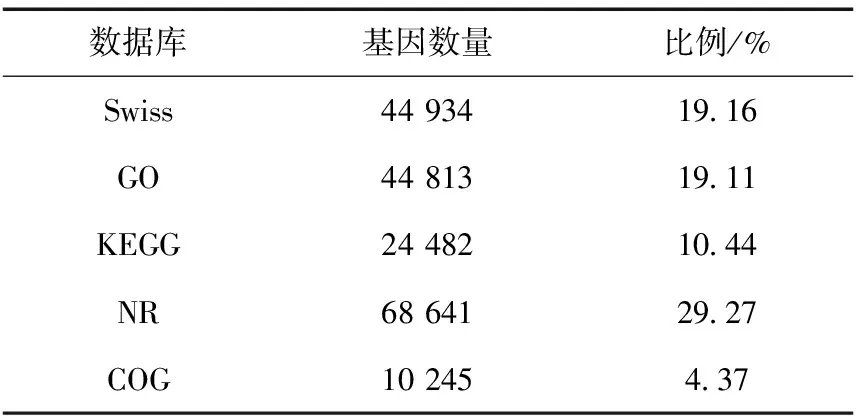
表3 五大数据库注释结果Table 3 Annotation results of five major databases
2.4 基因的GO注释结果
GO数据库将注释得到的基因分为细胞成分(cellular component)、分子功能(molecular function)和生物过程(biological process)三大类,在细胞成分大类中,细胞(cell)和细胞组分(cell part)注释到的基因最多;在分子功能大类中,结合(bingding)注释到的基因最多;在生物过程大类中,细胞过程(cellular process)注释到的基因最多(图2)。
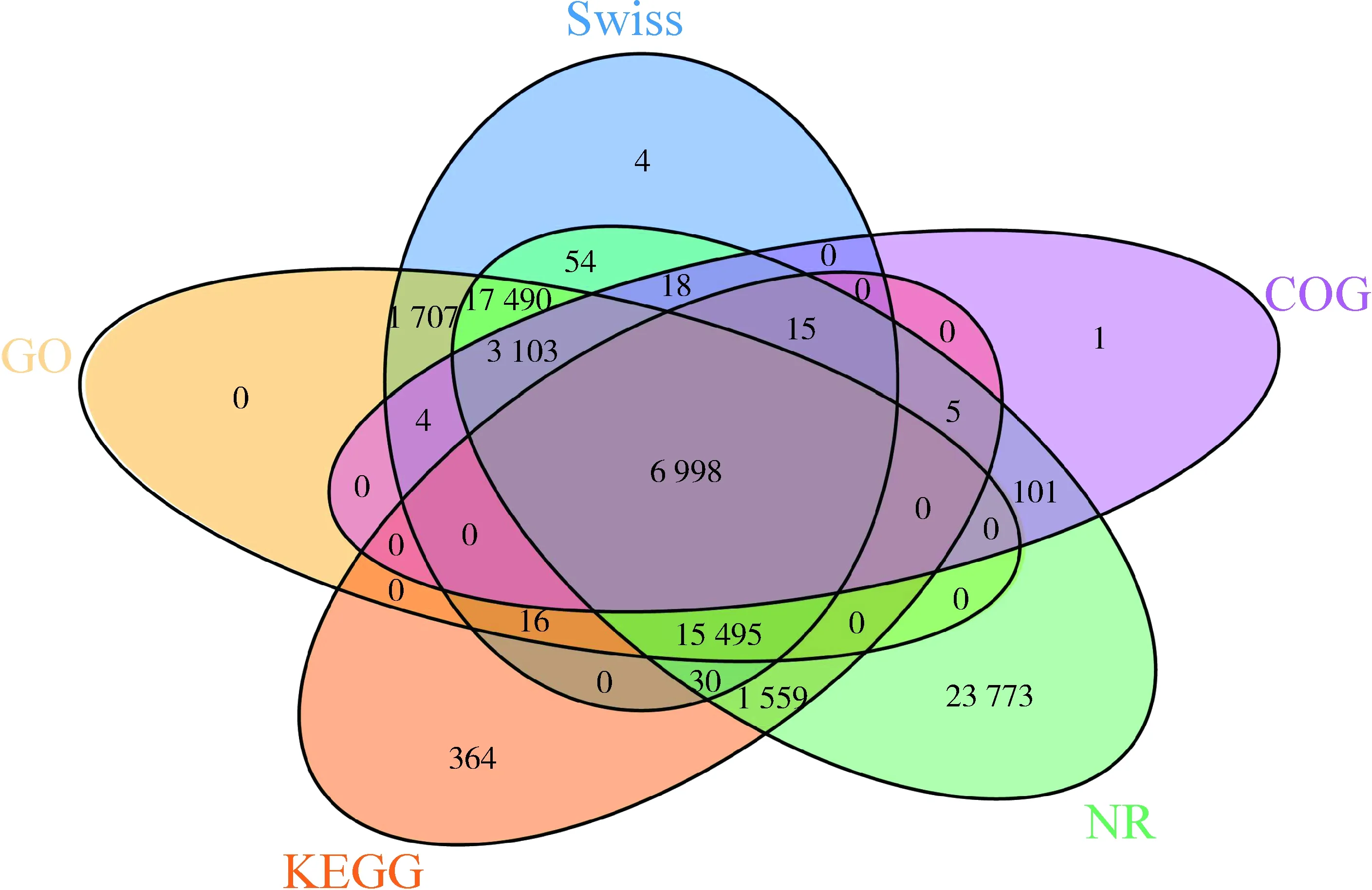
图1 四川山鹧鸪转录组组装的基因的数据库注释韦恩图Fig. 1 Venn diagram of database annotation of Arborophila rufipectus transcriptome assembled genes
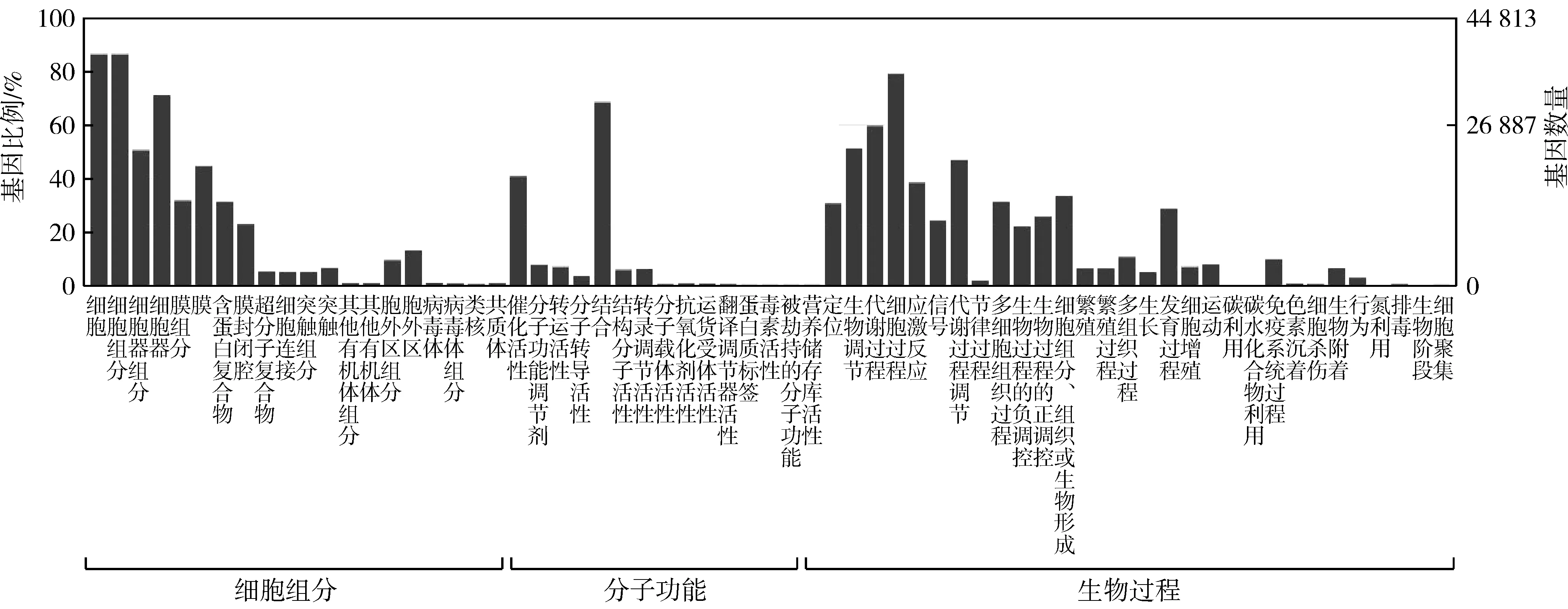
图2 四川山鹧鸪转录组GO数据库注释图Fig. 2 GO database annotation diagram of Arborophila rufipectus transcriptome
3 讨论
本研究采用读长短但准确性高的二代Illumina测序。数据质量控制检测报告显示,每条序列各位置碱基的测序质量均位于代表数据质量很好的绿色区间,每条序列各位置中无法识别的N碱基比例可以忽略不计,这体现了二代测序的准确性。但二代测序的读长短(50~500 bp),如基于150 bp的配对末端测序长度进行测序组装出来的基因的Contig N50只有964 bp,说明组装出来的基因碎片化,从大量的短序列中准确组装出完整的转录组仍然充满挑战。三代测序解决了读长问题,其测序长度高达上百kbp,但错误率高(1%~15%),需要二代测序辅助降低错误率。
从头组装的方法是基于序列之间的重叠部分完成的转录组组装,使用从头组装的方法进行组装的应用价值是对四川山鹧鸪原有基因组的补充,因其已有参考基因组也是通过二代Illumina测序组装的(Zhou.,2019),其组装过程中存在错误和组装不完全的情况。相对于基于DNA的基因组组装来说,基于RNA的可以将那些由多个转录本加工而成的成熟mRNA组装出来。在研究目标是转录本序列,且物种无参考基因组或者参考基因组质量不高的转录组组装中,Trinity是所有从头组装软件中准确度最高且组装更完整的软件,能在不损失准确性和运行速度的前提下得到最完整的组装版本(卢戌,2013)。
基因的注释是整个流程中最重要的一个环节,尤其是在高通量测序成本日益下降的情况下。组装拼接出来的草图需要进行注释后才能体现价值,后续的功能基因挖掘也建立在注释结果上。基因功能的注释是建立在结构注释的基础上的,根据结构注释的结果提取具有翻译功能的区域,并与主要的数据库比对。结构注释中可以看到只有18.71%的基因具有ORF框,即这部分序列有编码翻译成蛋白的潜能。这种现象普遍存在于真核生物的基因组中,大部分的基因被认为是“垃圾”序列,但具有重要的转录后调控作用,涉及转录起始、转录中和转录后的每一个过程,如lncRNA、MicroRNA、piRNA、siRNA等(Kapranov.,2007)。功能注释中NR数据库的结果最丰富。
四川山鹧鸪的近缘物种海南山鹧鸪的栖息地主要是海南岛的热带雨林和山地常绿林。海南山鹧鸪在海南霸王岭国家级自然保护区海拔350~1 560 m(韦锋等,2008)、海拔80~260 m的南味岭林区的次生林均有分布(邱胜荣,丁长春,2014)。四川山鹧鸪在四川麻咪泽自然保护区内的生境(2 100~2 300 m)(赵成等,2015)与海南山鹧鸪有明显的海拔梯度差异,后续可利用本研究数据与海南山鹧鸪进行比较转录组学研究,探讨近缘物种对不同海拔生境适应的遗传学机制。
猜你喜欢
戏曲研究(2022年3期)2022-05-10 08:09:44
扬子江诗刊(2022年1期)2022-01-10 05:43:28
扬子江(2022年1期)2022-01-07 19:26:38
今日农业(2021年11期)2021-08-13 08:53:24
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
金山(2016年1期)2016-08-17 22:03:34
遗传(2014年3期)2014-02-28 20:58:49
