
SCN5A基因突变致扩张型心肌病伴房室传导阻滞的家系调查
2022-01-20 01:41孙爱军葛均波
中国临床医学 2021年6期
徐 磊, 申 程, 孙爱军*, 葛均波
1. 复旦大学附属中山医院心内科,上海市心血管病研究所,上海 200032 2. 济宁医科大学附属医院心内科,济宁 272000
扩张型心肌病(dilated cardiomyopathy,DCM)主要表现为心腔扩大及收缩功能下降引起的心脏收缩功能减退,是心力衰竭及心脏移植的常见病因[1],患病率超过0.04%[2],1年内死亡率高达30%、5年生存率仅50%[1]。随着分子遗传学的发展,已发现超过100个基因与DCM有关[3]。在DCM患者中,家族性DCM占25%~30%,建议家族性DCM的所有家系成员行基因检测[4]。目前发现与家族性DCM有关的基因超过60个,高频致病基因主要包括肌联蛋白Titin(TTN)、核纤层蛋白 A/C(LMNA)、β肌球蛋白重链7(MYH7)、肌质RNA结合基序蛋白20(RBM20)、心脏钠离子通道α亚基(SCN5A)等[5]。
SCN5A基因编码心脏钠离子通道,而心脏钠离子通道是心肌细胞动作电位的重要组成部分,因此最早关于SCN5A突变的报道常与心律失常有关[6]。以往认为SCN5A通过致心律失常间接引发心脏扩大。2008年本课题组[7]在一DCM伴房室传导阻滞(atrioventricular block, AVB)的家系中发现了SCN5A的一个新突变A1180V,并观察到A1180V突变可直接影响心肌细胞内钙稳态而损伤心肌,导致心脏扩大。这一发现为SCN5A直接引起DCM提供了证据。同时,本课题组[7-8]还发现A1180V突变携带者表现出心率依赖性的心电图参数异常,这与A1180V突变型钠通道的电生理异常相一致,提示心率增加可能会促进突变携带者疾病表型出现。
2012年随访该家系时发现,尽管第3代成员中仅有1例突变携带者出现Ⅲ度AVB,仍建议所有突变携带者尽量避免劳累或从事重体力劳动,避免心率过快[9]。本次随访距最初发现该家系已14年,本次随访旨在进一步探讨SCN5A基因A1180V突变导致DCM的致病规律并找寻防治策略。现将随访情况报告如下。
1 资料与方法
1.1 一般资料 参考2007年和2012年该DCM伴AVB家系成员的临床资料及测序结果[7,9],经复旦大学附属中山医院伦理委员会审核批准(B2021-382R),取得所有健在对象的同意并签署知情同意书后,于2021年对该家系中健在的6名A1180V突变携带者和7名非突变携带者再次随访,予以询问病史、体格检查、12导联心电图及超声心动图检查。同时回顾性分析该家系中10名突变携带者(包括3名已故突变携带者),7名非突变携带者及1名已故基因型不详成员的临床资料,了解A1180V突变所致DCM的病情进展特点。随访该家系共3代,主要为先证者及其一、二级亲属:其父亲及子女3人,同父母的2个胞弟和1个胞妹及各自的子女。
1.2 诊断标准 DCM的诊断参考2018年《中国扩张型心肌病诊断和治疗指南》[5],以超声心动图为诊断依据。(1)心脏扩大:早期左心室扩大,后期各心腔均扩大,常合并二尖瓣和三尖瓣反流、肺动脉高压;(2)左室壁运动减弱:绝大多数左室壁运动弥漫性减弱、室壁相对变薄,可合并右室壁运动减弱;(3)左室收缩功能下降:左室射血分数(left ventricular ejection fraction, LVEF)<45%、左室短轴缩短率(left ventricular fractional shortening, LVFS)<25%,合并右室收缩功能下降时,三尖瓣收缩期位移(tircuspid annular plane systolic excursion,TAPSE)<1.7 cm、右室面积变化分数(fractional area change,FAC)<35%;(4)其他:附壁血栓多发生在左室心尖部。
AVB的诊断参考第9版《内科学》的诊断标准,以心电图为诊断依据。Ⅰ度AVB:每个P波后均有QRS波,PR间期>0.20 s。Ⅱ度Ⅰ型AVB:(1)PR间期进行性延长,直至一个P波下传受阻;(2)相邻RR间期进行性缩短,直至心室波脱漏;(3)包含受阻P波在内的RR间期小于正常窦性PP间期的2倍。Ⅲ度AVB:(1)心房心室活动各自独立;(2)心房率快于心室率;(3)心室起搏点多位于阻滞部位稍下方。
2 结 果
2.1 先证者及已死亡家庭成员临床资料 先证者(Ⅱ-1,A1180V携带者):37岁因心悸不适首诊为Ⅰ度AVB,超声心动图结果无异常;41岁超声心动图提示心脏扩大,伴发Ⅲ度AVB和房颤;47岁时安装起搏器,但心功能仍继续恶化,55岁因严重心力衰竭死亡。先证者父亲(Ⅰ-2,基因型不详):约35岁时出现心悸、气短等不适,体力活动受限,47岁心电图提示Ⅲ度AVB,51岁时因“心脏病”(具体不详)死亡。先证者弟弟(Ⅱ-3,A1180V携带者):37岁首诊为Ⅰ度AVB,40岁进展为Ⅲ度AVB,约5年后发展为DCM伴发Ⅲ度AVB,53岁时因心衰死亡。家系图见图1。
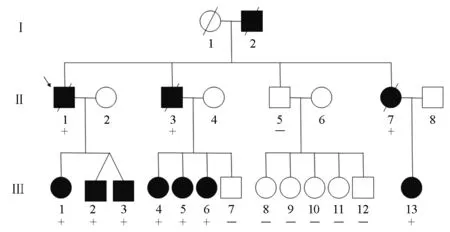
图1 扩张型心肌病伴房室传导阻滞家系图 “↓”为先证者;“﹢”为A1180V突变携带者;“-”为无突变者;“/”为已死亡。方框为男性;圆圈为女性。
2.2 家系其他A1180V突变携带者随访资料 A1180V突变携带者Ⅱ-7、Ⅲ-1、Ⅲ-2、Ⅲ-3、Ⅲ-4、Ⅲ-5、Ⅲ-6、Ⅲ-13在随访14年期间,疾病表型逐渐出现并加重。
先证者妹妹(Ⅱ-7)37岁初诊为Ⅰ度AVB,41岁进展为Ⅲ度AVB,常伴一过性黑朦;2007年(53岁)发现心脏扩大;2012年(58岁)发现病症进一步加重。自诉常感胸闷乏力、体力活动明显受限。体格检查发现心脏扩大,心电图示Ⅲ度AVB,超声心动图示全心扩大、二尖瓣和三尖瓣中重度反流、中度肺动脉高压、LVEF仅为36%。2012年随访时建议其尽快植入起搏器,但由于其顾虑费用一直拖延治疗,于当年猝死。
先证者女儿(Ⅲ-1)31岁时表现为Ⅰ度AVB;35岁时未发现心脏扩大;2012年(42岁)随访自诉偶感不适,体力活动轻度受限,心电图示Ⅲ度AVB,超声心动图提示左心扩大、多瓣膜轻度反流;2019年(49岁)因房扑伴Ⅲ度AVB植入单腔起搏器;2021年(51岁)随访时,心电图示起搏心律呈VVI模式,心率60 次/min,超声心动图提示心房稍大、室壁收缩活动正常、LVEF为66%。
先证者双胞胎儿子(Ⅲ-2、Ⅲ-3)2007年经病史询问和体检均未发现明显异常;2012年(33岁)均自诉偶感心悸不适,重体力劳动受限,心电图示Ⅰ度AVB,超声心动图示三尖瓣微量反流。Ⅲ-2于2020年(41岁)诊断为Ⅲ度AVB,行双腔起搏器植入术;2021年(42岁)随访时心电图为窦性心律、VAT模式起搏,心率70 次/min,超声心动图示左室临界大小、室间隔增厚(14 mm)、室壁收缩活动未见异常、LVEF为65%。Ⅲ-3于2018年(39岁)诊断为Ⅲ度AVB,行双腔起搏器植入术;此次随访时,心电图示窦性心律、VAT模式起搏、心率73次/min,超声心动图提示左房临界大小、室间隔增厚(13 mm)、室壁收缩活动未见异常、LVEF为62%。
先证者弟弟(Ⅱ-3)的三位女儿(Ⅲ-4、Ⅲ-5、Ⅲ-6)均为突变携带者,在2012年随访时均无不适症状,心电图及超声心动图检查无异常。Ⅲ-5于2015年(38岁) 发现Ⅰ度AVB,未予重视,近年体力劳动多。此次随访主诉乏力症状明显,心电图示已发展为Ⅲ度AVB,起搏器尚未植入,建议患者尽快行起搏器植入术;超声心动图提示双房增大,轻度二尖瓣、三尖瓣反流,室壁收缩活动未见异常。此次随访时,Ⅲ-4(47岁)和Ⅲ-6(39岁)虽无不适主诉,但心电图均出现Ⅰ度AVB,PR间期长达300 ms,建议其密切随访心电图;超声心动图仅见轻度二尖瓣、三尖瓣反流,心脏大小及功能均正常。
先证者妹妹(Ⅱ-7)的女儿(Ⅲ-13)25岁时心电图提示PR间期处于临界值;2012年(30岁)随访进展为Ⅱ度Ⅰ型AVB,超声心动图无异常表现。但该成员未能参加此次随访。
2.3 携带者总体表现 分析该家系所有A1180V突变携带者(包括已死亡的Ⅱ-1、Ⅱ-3和Ⅱ-7)的临床特点(表1)发现,A1180V突变携带者常因心悸乏力、运动耐受力下降就诊,接诊时心电图表现为Ⅰ度AVB,平均诊断年龄为(36±4.42)岁,此后病情多逐渐加重,进展为心脏扩大伴Ⅲ度AVB。但是,2012年至2021年,携带者出现Ⅲ度AVB和DCM的趋势在延缓。
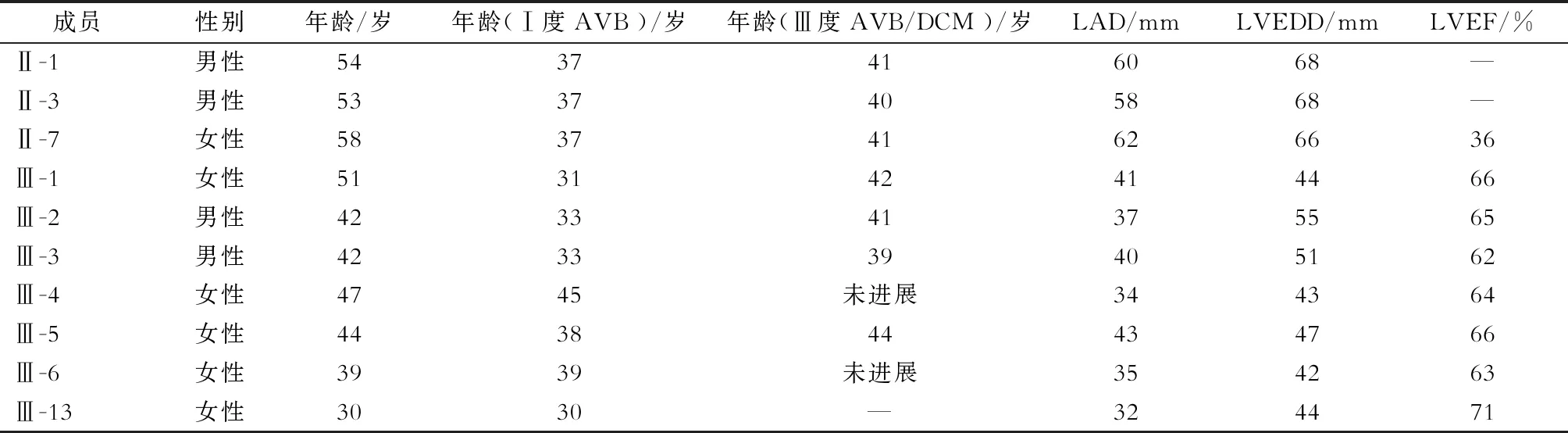
表 1 该家系中A1180V突变携带者的此次随访资料
2.4 A1180V突变非携带者情况 A1180V突变非携带者Ⅱ-5、Ⅲ-7、Ⅲ-8、Ⅲ-9、Ⅲ-10、Ⅲ-11、Ⅲ-12经询问病史、体格检差、心电图及超声心动图等均未发现明显异常。
3 讨 论
3.1 A1180V突变携带者病情进展差异原因 虽然随着年龄增长,A1180V突变携带者的症状逐渐出现并加重,多在约36岁诊断为Ⅰ度AVB,随后逐渐进展为Ⅲ度AVB伴心脏扩大,但是体力劳动程度影响其进展。Ⅲ-1作为第3代的最长者,平素性格温和,不喜劳作,此次随访时已51岁,虽然发现Ⅲ度AVB的年龄与长辈相似,但是此次随访时其心脏收缩功能仍未见明显减弱且左室不大。而其父亲在41岁时即有心脏扩大和心衰表现,说明出现DCM前及时植入起搏器改善Ⅲ度AVB、避免重体力劳动和注意控制心率使该携带者出现DCM的年龄较长辈至少延后10年。Ⅲ-4、Ⅲ-5、Ⅲ-6三姐妹中Ⅲ-5的病情发展最快。询问病史发现,Ⅲ-5平时参与重体力劳动多。另外,虽然Ⅲ-13此次未能参加随访,但2012年随访时发现该成员常参与体育比赛项目,其心电图出现异常的年龄要明显早于其他携带者。因此,对于A1180V突变携带者,后天通过控制心率、避免情绪激动和避免重体力劳动能有效延缓疾病的发生和进展。此次随访过程中,反复对突变携带者强调控制心率和避免重体力劳动的重要性。
先证者的3位子女(Ⅲ-1、Ⅲ-2、Ⅲ-3)均已出现Ⅲ度AVB并及时植入起搏器,虽然3人均已超过其父发生DCM的年龄,但此次随访未发现心脏功能异常,日常生活中未出现相关不适,而其父在植入起搏器后心脏功能仍持续恶化并死亡。这种表现的原因可能为:(1)先证者植入起搏器时已出现DCM表型,贻误了治疗时机,而其3位子女植入起搏器时心脏功能均正常;(2)3位子女已遵医嘱,不再从事重体力劳动,避免劳累。因此,对突变携带者定期随访、发现问题及早干预、避免重体力劳动是延缓出现DCM表型的重要策略。控制心率和避免重体力劳动对于DCM患者均适用,而对于该家系携带者,这一措施应在其没有出现心脏扩大、甚至没有出现AVB前就进行预防,即A1180V突变的年轻携带者应尽早控制心率。该策略符合A1180V突变的电生理特性[7]。
3.2 SCN5A突变致心脏功能障碍机制 目前已发现多个SCN5A突变和DCM有关,如R225W、R225P、R814W和R219H等[10-12]。虽然SCN5A突变对DCM的致病性已明确,但具体机制目前仍未明确。除了A1180V突变导致钙稳态失衡引起DCM外[7],本课题组在散发性DCM患者中发现,SCN5A的另外3个错义突变(R225Q、A226V和I1448N)编码的蛋白均位于心脏钠通道电压感受器结构域Ⅰ的S4部位(DⅠ-S4)。通过膜片钳技术研究发现,突变的钠通道中峰值钠电流密度减小,进而导致心肌细胞受损[13]。另有研究[14]发现,SCN5A突变可产生阳离子泄露的门孔电流,这种泄露电流与心律失常及心脏扩大有关。上述研究提示,SCN5A突变引起的心脏钠通道功能减弱或增强均可能引起DCM。功能减弱的突变钠通道可能通过干扰细胞骨架蛋白结合导致心肌收缩活动减弱,同时通过引起心肌细胞电机械耦联异常导致心脏扩大;功能增强的突变钠通道主要影响浦肯野系统,使浦肯野细胞复极化不完全,使心室动作电位过早出现,进而导致DCM[15-16]。不同SCN5A突变致DCM的具体机制还需要通过构建相应的转基因动物模型来进一步分析。
此外,先证者的3位子女(Ⅲ-1、Ⅲ-2、Ⅲ-3)和先证者胞弟的3位女儿(Ⅲ-4、Ⅲ-5、Ⅲ-6)同为A1180V突变携带者,但先证者3位子女AVB表型出现较早,提示母亲的遗传因素可能对疾病发展发挥作用,也可能存在寡基因(主效基因和微效基因)的互相作用。2019年,Science杂志报道的一个寡基因遗传导致左室致密化不全心肌病家系中,先证者父亲携带MYH7和MKL2两个突变且有表型,母亲携带NKX2-5突变,父母均无症状,但先证者和其2个同胞均出现心脏发育异常[17]。DCM是否存在类似的寡基因遗传致病机制目前尚鲜见报道。本课题组随访的A1180V突变家系中,第3代是否受到母亲基因的影响而使来自父亲A1180V突变的表型加重或减弱尚不明确,还需要采取更详尽的全基因组或全外显子测序方法进一步分析。
综上所述,本研究通过对该家系长达14年的随访,进一步证实了A1180V突变的致DCM作用。随访结果提示,定期行心电图和超声心动图检查有助于发现A1180V突变携带者的早期病变;避免重体力劳动及情绪激动,Ⅲ度AVB患者在尚未发生心衰前尽早干预并植入起搏器,有助于延缓甚至预防疾病进展。本课题组会对该家系成员继续进行随访,追踪年轻携带者是否会出现DCM表型,以深入探讨A1180V突变与DCM表型的关系。
利益冲突:所有作者声明不存在利益冲突。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
园艺与种苗(2021年8期)2021-09-23
中老年保健(2021年12期)2021-08-24
心电与循环(2021年1期)2021-02-05
幸福家庭(2020年17期)2020-12-10
保健与生活(2020年4期)2020-03-02
健康博览(2020年1期)2020-02-27
森林工程(2018年1期)2018-05-14
