
Ni-Mo复合金属催化剂在几种杂环伯胺中的合成
2022-01-20 10:59张传保冯柏成王永琪
青岛科技大学学报(自然科学版) 2022年1期
张传保,冯柏成,王永琪
(青岛科技大学 化工学院,山东 青岛 266042)
伯胺被认为是化学领域中的重要基石,已广泛用于聚合物、染料、颜料、农药、药物、表面活性剂等[1-3]。伯胺也是最有用的中间体之一,现在可以通过多种方式合成伯胺,例如腈和硝基化合物的还原[4-5]、卤化物与氨的烷基化[6-7]、醇[8]和羰基化合物[9]的胺化。其中,羰基化合物与氨气和氢气的还原胺化是最合适的合成相应伯胺的方法之一,由于水是唯一的副产物,因此伯胺的原子效率高且对环境友好。关于杂环伯胺,其不仅应用在医药行业,而且在高分子聚合物中,特别是在聚酰亚胺膜中也有着广泛的应用。有研究发现在液晶取向剂的合成中引入杂环伯胺的结构能提高取向剂的取向性能,从而进一步提高了液晶膜的品质,因此研究者们正在逐步地开发和应用这种取向剂[10-11]。
目前,合成伯胺主要有两种方法。一种是将羰基化合物还原为伯胺,该方法主要分两步合成[12]:(1)羰基化合物与氨缩合脱水形成亚胺;(2)通过催化氢化将亚胺还原得到伯胺;另一种方法是将醛转化为肟,然后通过催化氢化将肟还原成伯胺[13]。实际上,与第二种方法相比,前者具有经济上的优势,但此方法所获得的收率不稳定,不适合规模化生产,究其原因主要是由于亚胺中间体不稳定,可能自凝结形成二聚,甚至三聚体形式[14],而第二种方法虽然能得到较稳定的中间体,但是在产品收率和选择性上又不太令人满意。因此,对第二法又做了进一步研究及改进。
研究发现在醛转化为肟的反应中,压力、温度和时间对产率虽然有影响,但是溶剂中NH3浓度是影响产物分布、提高收率的关键。溶剂中NH3可以有效地抑制仲胺和叔胺的生成,提高选择性。在还原过程中,NH3的抑制能力不仅优于H2O,而且溶于溶剂中的NH3比直接通氨气可以发挥更好的性能[15-16]。
另外,大量的研究表明,在催化氢化过程中,以金属为基底的均相催化剂如钌基和铱基的络合物[17-20]以及金属催化剂Pt、Ru、Pd、Co、Ni、Cu等可以还原合成胺[21-26]。此外,金属氢化物如Li-Al H4[27-28]或NaBH4[29]也常作为还原剂。但是由于大多数金属催化剂具有湿敏性和高成本,所以不太适用于大规模及杂环化合物的生产。
为改进上述问题,本研究将之前添加NH3·H2O或通入带压NH3改为直接使用预先制备的氨甲醇溶液作反应溶剂,该操作可进一步提高产品的选择性和收率。同时改进了常用的Ni催化剂,并通过自制的Ni-Mo金属催化剂还原杂环,结果表明自制Ni-Mo金属催化剂比常规催化剂Raney Ni在还原过程中对产物的收率和选择性更有优势。
1 实验部分
1.1 材料与仪器
2-呋喃甲醛、2-噻吩甲醛、3-吡啶甲醛、2-吡啶甲醛、甲醇,国药集团化学试剂;Pd/C,Pt/C,陕西瑞科新材料股份有限公司;Raney Ni,辽宁众力催化剂科技有限公司。
数字熔点仪,WRS-2型,上海易测仪器设备有限公司;核磁NMR(400 MHz),Bruker Avance型,瑞士Bruker公 司;X射 线 衍 射 仪,“X”Pert PROMPD型,荷兰Nalytical公司;扫描电镜,JSM-6510A型,日本电子公司。
1.2 Ni-Mo催化剂的合成
将六水硝酸镍和四水合钼酸铵按照一定物质的量比例溶解于一定量的去离子水中搅拌至完全溶解,配成一定浓度的溶液。再加入一定量的络合剂一水合柠檬酸,络合剂物质的量为金属阳离子物质的量的1.2倍,待完全溶解后,于80℃下搅拌至凝胶状。将凝胶状催化剂在110℃下干燥10 h。将干燥好的固体迅速放置在马弗炉中于550℃下煅烧6 h,再将其于0.1~2.0 MPa氢气压力下还原得,得Ni-Mo催化剂。
1.3 肟的合成
向一定量的溶剂中加入杂环醛和盐酸羟胺,再逐渐滴加碳酸钠(醛、盐酸羟胺和碳酸钠的物质的量的比1∶1.1∶0.55),加完后,在连续搅拌下从室温缓慢加热至回流1~3 h。然后冷却,过滤,滤饼用水洗涤,无需纯化可直接用于下一步。
1.4 肟的还原胺化
将一定浓度的氨甲醇溶液600 m L、60 g醛肟和6 g催化剂加入1 000 mL不锈钢高压釜中。氢气置换3~5次,以1 000 r·min-1连续搅拌。反应后,将高压釜冷却至室温并取样通过GC分析,分析条件为色谱柱的初始温度为80℃,并保持2 min,以20℃·min-1的速率将温度升至270℃,保持5 min。
2 结果与讨论
2.1 产品的表征
2-呋喃甲胺(1b),1H NMR(400 MHz,DMSO,δ):7.51(s,1H),6.36(dd,J=3.1,1.9 Hz,1H),6.19(dd,J=3.1,0.9 Hz,1H),3.66(s,2H)。13C NMR(101 MHz,DMSO,δ):157.87(s),141.75(s),110.73(s),105.32(s),39.81(s)。
2-四氢呋喃甲胺(1c),1H NMR(400 MHz,CDCl3,δ):3.87(dd,J=6.6,2.5 Hz,1H),3.76(dd,J=14.1,7.4 Hz,1 H),3.13~2.52(m,1H),1.90(ddd,J=20.1,12.5,7.7 Hz,2 H),1.72~1.50(m,1 H),1.37(s,1H)。13C NMR(101 MHz,CDCl3,δ):80.70(s),67.75(s),46.45(s),28.69(s),25.85(s)。
2-噻吩甲胺(2b),1H NMR(400 MHz,CDCl3,δ):7.18(dd,J=8.1,3.0 Hz,1H),7.03~6.85(m,2H),4.05(s,2H),1.60(s,2H)。13C NMR(101 MHz,CDCl3,δ):147.44(s),126.81(s),123.98(s),123.60(s),41.34(s)。
2-氨甲基吡啶(3b),1H NMR(500 MHz,DMSO,δ):8.48(d,J=4.5 Hz,1H),7.73(td,J=7.7,1.6 Hz,1 H),7.43(d,J=7.8 Hz,1 H),7.21(dd,J=6.8,5.4 Hz,1H),3.81(s,2 H)。13C NMR(126 MHz,CDCl3,δ):163.34(s),148.91(s),136.73(s),121.86(s),121.24(s),47.76(s)。
3-氨甲基吡啶(4b),1H NMR(400 MHz,CDCl3,δ):8.61~8.01(m,1H),7.65(t,J=9.6 Hz,1H),7.23(dd,J=12.1,7.2 Hz,1H),3.92~3.43(m,1H),1.56(s,1H)。13C NMR(101 MHz,CDCl3,δ):148.88(s),148.20(s),138.21(s),134.47(s),123.37(s),43.50(s)。
2.2 催化剂的筛选
将六水硝酸镍和四水合钼酸铵分别按照金属镍钼的物质的量的比为1∶1,2∶1,4∶1的比例溶解于一定量的去离子水中,配成浓度约为30%的溶液。再加入金属阳离子1.2倍物质的量的一水合柠檬酸作为络合剂,待完全溶解后,于80℃下搅拌至凝胶状。将凝胶状催化剂在110℃下干燥10 h。将干燥好的固体迅速放置在马弗炉中于550℃下煅烧6 h,得NiMoOx复合氧化物。并通过XRD进行了晶体分析,结果见表1。镍钼的物质的量比为1∶1和4∶1的催化剂均为混合晶体,而物质的量的比2∶1为单晶,因此选择物质的量的比2∶1为催化剂合成比例。

表1 XRD晶型结构分析Table 1 Crystal structure analysis of XRD
2.3 催化剂的表征与应用
2.3.1 XRD和SEM分 析
图1是Ni-Mo复合氧化物的X射线衍射图。可 以 看 到,在2θ为25.3°、26.5°、28.4°、32.2°、36.7°、43.6°和47.5°出现明显的衍射峰,依次与Ni-Mo O4的标准卡片(PDF#33-0948)的(-112)、(-301)、(220)、(130)、(-231)、(330)和(-204)的晶面相吻合,而且图中并没有出现其他明显的衍射峰,表明催化剂纯度高。图2显示了催化剂表面光滑,呈较规则的棒状或者长条状,无明显的团聚现象,比表面积较大,说明催化剂径向表面活性大,有利于物质的扩散和加氢反应的进行。

图1 Ni-Mo复合氧化物的X射线衍射图Fig.1 XRD spectrum of Ni-Mo catalyst

图2 Ni-Mo复合金属催化剂SEM照片Fig.2 SEM image of Ni-Mo catalyst
2.3.2 XPS分析
采用X射线光电子能谱(XPS)检测催化剂中元素组成和价态,见图3。如图3(a)所示,Ni 2p光谱在结合能峰在856.0 eV及其附属峰861.9 eV处与Ni 2p3/2相对应,而在873.8和880.1 eV的峰归于Ni 2p1/2。Ni 2p3/2和Ni 2p1/2的峰之间的分离能为17.8 eV,是Ni2+氧化态。同时,通过对比Ni-Mo催化剂和Raney Ni的Ni 2p光谱发现俩者并没有明显差异,说明金属Mo的引入对Ni的价态没有影响。如图3(b)所示,Mo 3d5/2和Mo 3d3/2峰的结合能分别为232.6和235.7 eV,分裂能为3.1 e V。这些特性与Mo6+的氧化态一致。此外,确认228.3和231.7 eV处是Mo 3d5/2和Mo 3d3/2的两个峰,这是Mo4+氧化态。这些XPS结果证明了Ni元素的化合价分别为+2,而Mo元素的化合价为+4,+6。
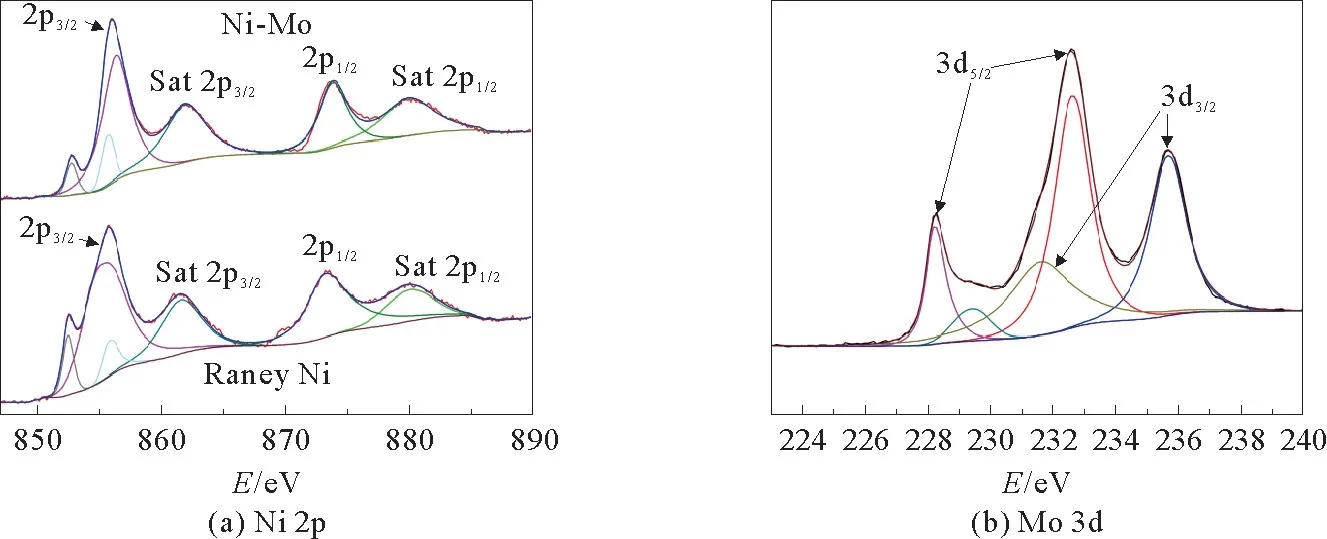
图3 Ni-Mo和Raney Ni的XPS光谱Fig.3 XPS spectra of Ni-Mo and Raney Ni
2.3.3 BET低温氮气吸脱附
表2、图4和图5为催化剂的BET分析及其分布图。从图表中看出,催化剂具有中孔结构。图4中,Ni-Mo催化剂的比表面积为33.2 m2·g-1比Raney Ni的28.7 m2·g-1高。此 外,Ni-Mo和Raney Ni孔径分布中心分别在31.5和40.9 nm处(图5),两者在孔径分布上相差不大,但是Ni-Mo却有较大的孔体积,其孔体积为0.15 cm3·g-1,而Raney Ni为0.12 cm3·g-1。分析结果表明,Ni-Mo催化剂将比Raney Ni有更大的比表面积,会是反应更有效,因为它可以提供更多的活性位点来促进化学反应。

表2 催化剂的BET性质Table 2 BET property of catalysts

图4 样品的恒温氮气吸附-脱附Fig.4 Nitrogen adsorption-desorption isotherm of samples

图5 样品的孔径分布Fig.5 Pore size distribution of samples
2.3.4 催化剂的应用
以2-糠醛甲肟还原成糠胺的还原胺化反应为例,来评估商业购买催化剂Pd/C,Pt/C,Raney Ni和自制Ni-Mo的催化活性,见表3。由表3发现,所用的催化剂在常压氢气下均没有使原料转化(如Pt/C,Raney Ni,Ni-Mo)(序号5,7,9)或着很低的转化率(如Pd/C)(序号1)。当引入1.0 MPa的氢气压力后,如所预期的一样,原料完全转化。然而,Ni-Mo催化剂相比其他金属催化剂得到了更高的伯胺(糠胺)收率(序号4,6,8,10)。通常Pd/C具有较高的加氢活性,因此通过改变氢气压力和减少Pd/C的使用量来进行实验,发现Pd/C对糠胺的选择性较低(序号2,3)。与Pd/C相比,在相同的还原条件下,Ni-Mo和Raney Ni对糠胺则表现出高的选择性。同样在相同的条件下,Ni-Mo和Raney Ni还原所得的糠胺收率分别为85.7%和90.5%,环氢化副产物(四氢呋喃甲胺)的收率分别为11.4%和5.7%,表明Ni-Mo比Raney Ni在环氢化产物上有更高的选择性,这说明相同条件下Ni-Mo催化活性更高(序号8,10)。因此,通过进一步优化反应条件,Ni-Mo表现出最佳的催化性能,糠胺收率达到最高的96.6%,仅有1.2%的环氢化产物和2.2%的其他副产物(序号12)。换句话说,Ni-Mo是在当前反应条件下用于还原胺化的最佳的催化剂。

表3 不同金属催化剂的还原反应Table 3 Reduction reaction of different metal catalysts
2.4 还原胺化反应
根据之前的研究,在还原过程中,选择了Ni-Mo复合金属作为催化剂,通过两个步骤由相应的醛合成了几种杂环伯胺。第一步中,将醛转化为相应的肟,值得注意的是,这些肟的合成是高效的,高收率的,结果见表4。
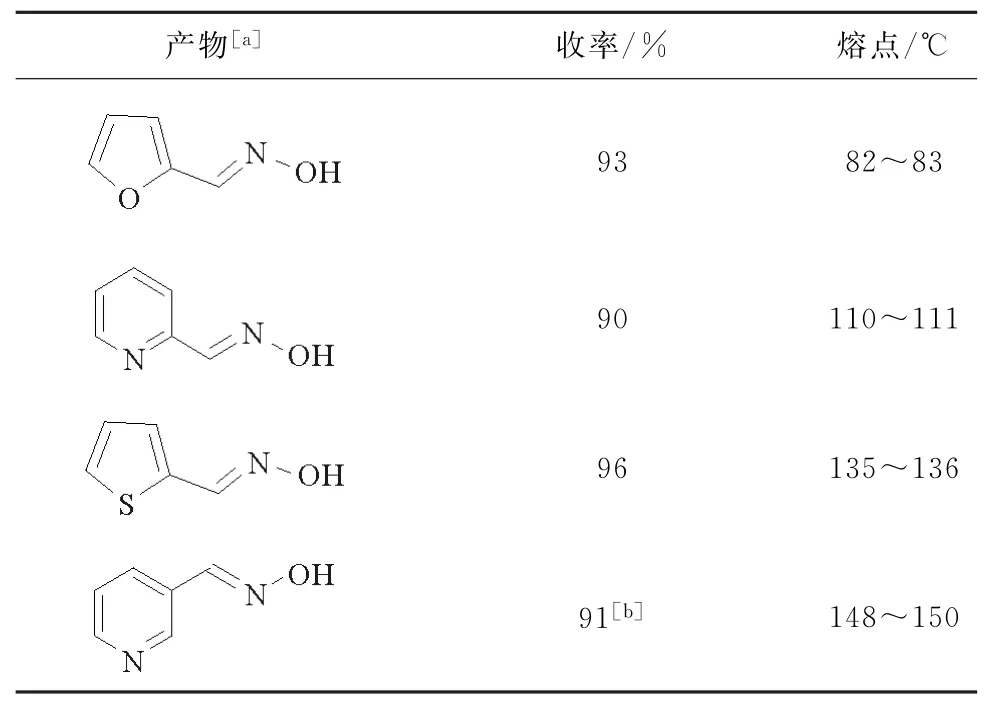
表4 不同杂环肟的合成Table 4 Synthesis of different heterocyclic oximes
第二步是将肟还原为相应的伯胺化合物,根据上述的方法,制备了一些代表性的杂环伯胺产品,证明了该方法是有效的,而且提供了具有良好收率,结果见表5。

表5 不同肟的还原反应Table 5 Reduction of different oximes
2.5 催化剂的再利用
从绿色化学和降低工业成本的角度出发,以糠胺的合成为例对Ni-Mo催化剂的可重复使用进行了研究。还原反应后,将催化剂过滤,用甲醇洗涤,再加到高压釜中在相同的反应条件下多次重复使用,结果见表6。催化剂在还原过程中保持了催化活性,可以重复使用至少4次且不会明显失活。当第5次应用时发现性能明显降低,除了回收过程中造成损失以外,另外一个原因可能是催化剂的活性位点,因此通过XPS分析了第5次与第1次催化剂中Mo原子的含量发现,第1次的为5.91%,而第4次的为0.5%。因此,对催化路线做了推测,反应过程中Ni是主要角色,溶液中的氢气吸附在Ni表面被还原成原子氢,使得Ni的化合价升高,而杂环肟同样吸附在Ni表面,与氢原子结合还原产生水,另外Mo4+的存在可能在反应过程中能快速将Ni2+还原,使得反应更加的高效[30-32]。
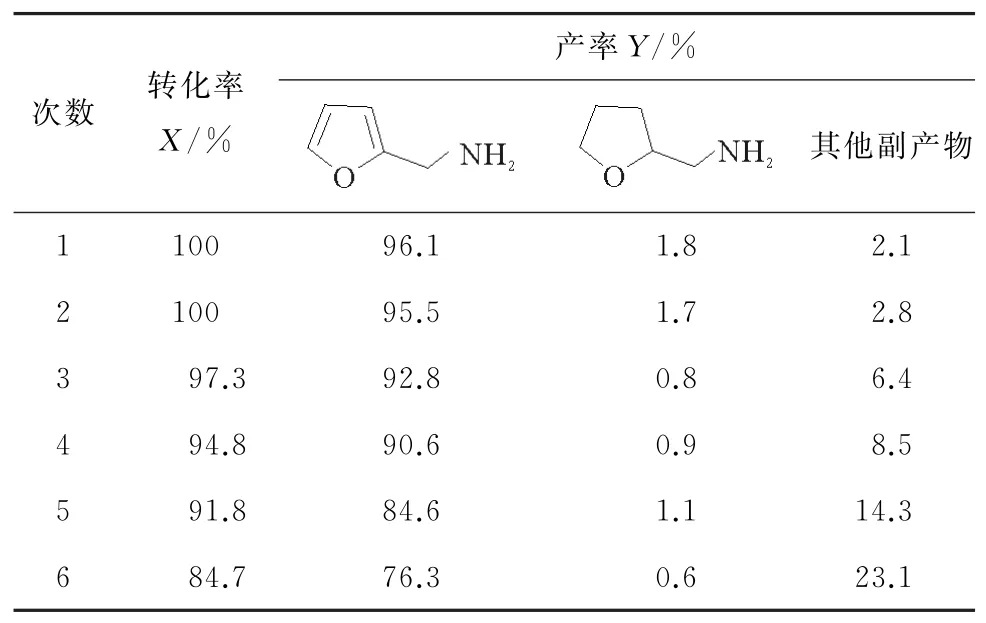
表6 催化剂的回收Table 6 Recovery of catalyst
3 结 论
通过X射线衍射(XRD)、X射线光电子能谱(XPS)、比表面积(BET)表征对合成的Ni-Mo复合金属催化剂进行了分析,并与常规催化剂Raney Ni做了对比,发现催化性能优于Raney Ni。同时,实验采用分步合成的方法,用合成的Ni-Mo催化剂还原肟获得相应的杂环伯胺,并获得96.6%的收率,且合成的杂环肟中间体稳定,操作方便。同时,也证明这种复合催化剂对杂环伯胺的还原具有显着影响,并且可以重复使用至少4次而不会明显失活。
猜你喜欢
石油炼制与化工(2022年10期)2022-11-26
世界农药(2022年10期)2022-11-10
分子催化(2022年1期)2022-11-02
云南化工(2022年2期)2022-03-18
建材发展导向(2021年16期)2021-10-12
农药科学与管理(2021年2期)2021-03-16
智富时代(2018年3期)2018-06-11
智富时代(2018年3期)2018-06-11
中国科技纵横(2016年13期)2016-08-22
Coco薇(2016年2期)2016-03-22
