
伯胺类化合物合成研究进展
2022-01-17 08:23龙奕华李汪涛王正宝
高校化学工程学报 2021年6期
俞 杰, 龙奕华, 李汪涛, 王正宝
伯胺类化合物合成研究进展
俞 杰1,2, 龙奕华1, 李汪涛1, 王正宝1
(1. 浙江大学 化学工程与生物工程学院, 浙江 杭州 310027; 2. 浙江树人大学 生物与环境工程学院, 浙江 杭州 310015)
伯胺类化合物是一类重要的有机化工原料,在医药、农药、食品添加剂、洗涤剂、润滑剂和功能高分子材料等领域均有十分广泛的应用。本文总结了卤代化合物与氨的烷基化反应、烯烃的氢化胺化反应、醇的氨解反应以及醛、酮化合物的还原胺化反应等通过催化反应合成伯胺的主要方法,着重介绍了各个方法所使用的催化体系,并简单介绍了相关反应机理。其中,醇的氨解反应和醛、酮化合物的还原胺化反应这两种方法具有较强的工业化前景,开发新型催化体系及其制备方法,从而有效提升催化剂的活性、选择性以及适用范围,将是该两种方法未来的主要发展方向。
伯胺;合成;催化;氨解;还原胺化
1 前言
胺类化合物,尤其是伯胺,是一类重要的精细化工原料,在医药、农药、食品添加剂、洗涤剂、润滑剂等领域均有十分广泛的应用[1-3]。此外,胺类化合物还可作为关键单体用于合成聚酰胺、聚酰亚胺、聚脲等功能高分子材料[4-6],进而用于生产汽车、航空和健康等领域的相关产品。因此,随着胺类化合物下游产品的不断开发,近年来,其需求量正逐步增加。
由于氮原子上的孤对电子易与质子结合,因此胺类化合物具有碱性。胺类化合物的碱性强弱主要与氮原子所连基团的性质有关。对于脂肪胺,烃基的给电子效应使得氮原子的电子云密度增高,结合质子的能力变强;但随着烃基数目增多,空间位阻变大,其结合质子能力减弱。此外,氮原子结合的氢质子越多,溶剂化能力就越强,碱性也越强。故脂肪胺的碱性是以上3种因素综合作用的结果,总体上,伯胺和仲胺比叔胺的碱性强。对于芳香胺,由于其氮原子上的孤对电子所占据的轨道和苯环的π轨道共轭,使得氮原子结合质子的能力减弱,故芳基越多,碱性越弱,因此,伯胺比仲胺和叔胺的碱性强。
研究结果表明[7],胺类化合物的亲核性能与其碱性正相关,而其亲核性能则反映了反应性能的高低。事实上,与仲胺和叔胺相比,伯胺氮原子上连有两个氢原子,而氢质子与其他基团相比易脱去,反应成键后还剩余一个氢原子可形成氢键,因此,在某种程度上,伯胺的结构特征更优。此外,一分子伯胺可交换两个氢质子,易与两分子环氧烷或丙烯酸酯反应生成交联产物,还可与环酐反应生成环状酰亚胺[8]等。因此,由于伯胺这种反应特性,引起人们极大兴趣。
经典的伯胺合成反应包括盖布瑞尔合成反应[9]、霍夫曼重排反应[10]、柯提斯重排反应[11]以及硝基化合物的还原反应[12]等。这些反应或步骤长、或反应条件苛刻、或原子经济性不高。在分子中直接引入NH2-官能团被认为是一项挑战性的任务[13]。在化学家们的努力下,使用氨水或其他氮源,通过催化官能团转换反应构筑伯胺类化合物的一系列工艺路线被开发出来。作者综述了近年来伯胺类化合物的主要合成方法,并初步分析了这些合成方法存在的问题,展望了未来的研究方向。
2 伯胺类化合物的主要合成方法
根据所用原料的不同,将近年来较为常见的通过催化官能团转换反应构筑伯胺类化合物的方法分为卤代化合物与氨的烷基化反应、烯烃的氢化胺化反应、醇的氨解反应和醛、酮化合物的还原胺化反应等合成方法。表1列出了这些合成方法的主要优缺点。

表1 伯胺类化合物主要合成方法优缺点比较
2.1 卤代化合物与氨的烷基化反应
脂肪卤代物与氨的烷基化制备伯胺的反应属于双分子亲核取代反应[14]。首先卤代烷与氨生成铵盐,然后弱酸性的铵盐与弱碱性的氨发生可逆的质子转移反应生成伯胺。伯胺的氮原子上仍有孤对电子,其亲核性通常比氨的氮原子强,进而可与卤代烷反应生成仲胺;仲胺可与卤代烷反应生成叔胺;叔胺又可与卤代烷反应生成铵盐。因此,这类反应的纯度和产率都不高,工业上通常只用于生产低级胺,且需通过蒸馏加以分离。
芳卤化合物的苯环与卤素共轭,故反应活性不高。要实现芳卤化合物与氨/胺的烷基化则需要应用Buchwald-Hartwig偶联反应,该反应是美国麻省理工学院的Buchwald等[15]和耶鲁大学的Hartwig等[16]在1994年提出的催化剂和碱存在下的氨/胺与芳卤的交叉偶联反应,是合成芳胺的重要方法。起初采用Buchwald-Hartwig偶联反应合成伯胺是以二苯甲酮亚胺[17]或N,N-二烯丙基胺[18]为氮源(图1),在Pd的催化和叔丁醇钠(-BuONa)作用下,先与卤代烷偶联,然后在酸性条件下裂解得到目标产品。

图1 以二苯甲酮亚胺或N, N-二烯丙基胺为氮源的Buchwald-Hartwig偶联反应[17-18]
2006年,Shen等[19]、梅苏宁等[20]报道了以氨为氮源直接合成芳基伯胺的Buchwald-Hartwig偶联反应,以Pd(CH3CN)2Cl2和二茂铁双膦配体CyPF--Bu (Josiphos配体)为原料,制备了一种能对氨烷基化取代稳定的 (CyPF--Bu)PdCl2催化剂,以叔丁醇钠为碱、1,2-二甲氧基乙烷(DME)为溶剂(图2),在各种卤代芳烃与氨的烷基化反应中都表现出很高的反应活性和伯胺选择性,伯胺的最高收率为94%。2007年,Surry等[21]以二亚苄基丙酮(三(Pd2(dba)3)二钯) 为Pd源,以叔丁醇钠为碱、1, 4-二氧六环为溶剂,并使用联苯膦配体,研究了卤代芳烃与氨的烷基化反应,伯胺的收率为60%~85%。随后,Schulz等[22]和Lundgren等[23]也考察了Pd催化下的该反应,反应活性和选择性均较高,伯胺的最高收率可达90%以上。Vo等[24]通过实验推测Pd催化下卤代芳烃与氨偶联反应的可能反应机理(图3):首先,从催化剂前体中释放出来的零价钯活性催化剂LnPd(0)与卤代芳烃氧化加成,形成二价钯的过渡态化合物,然后与氨配合形成催化剂-卤代芳烃-氨的过渡态配合物;该过渡态配合物在碱作用下脱去质子,形成钯-氨基配合物,再经还原消除后得到目标化合物芳香胺。
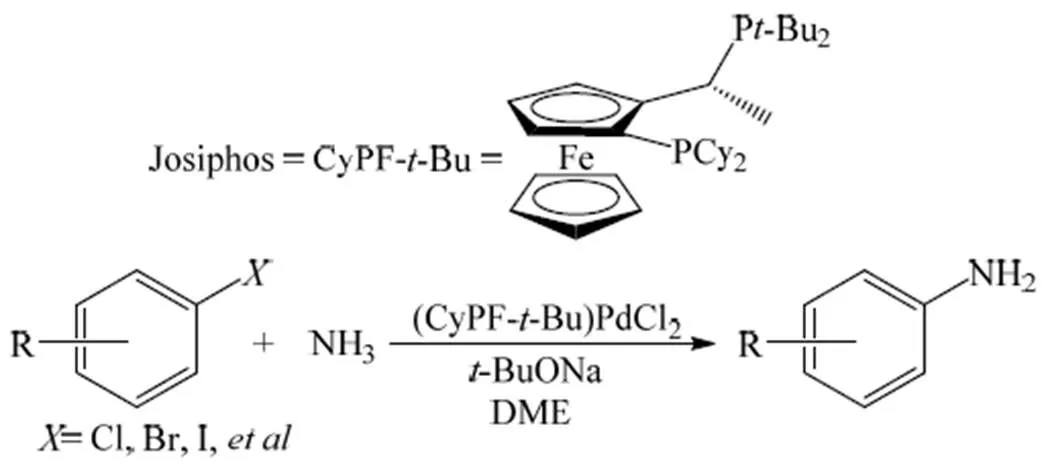
图2 Pd催化下以氨为氮源的Buchwald-Hartwig偶联反应[19]

图3 Pd催化卤代芳烃与氨偶联反应机理[224]
在非贵金属催化剂方面,Cu基催化剂是报道较多的催化剂 (图4)。2008年,Kim等[25]以CuI为催化剂、氨基酸为配体、K2CO3为碱、二甲基亚砜(DMSO)为溶剂,在室温下考察了芳卤与NH4Cl或氨的偶联反应,当芳环上连有供电子基团时,芳胺的收率相对较低,约为30%~80%;当芳环上连有吸电子基团时,芳胺的收率相对较高,可达80%以上。2013年,Fantasia等[26]以乙酰丙酮铜(Cu(C5H7O2)2)为催化剂、K3PO4为碱、N,N-二甲基甲酰胺(DMF)为溶剂,不使用配体,考察了杂芳溴与氮气的偶联反应,伯胺的收率为64%~88%。2019年,Abdine等[27]以CuBr为催化剂、吡啶基二酮为配体、K3PO4为碱,考察了芳卤与氨的偶联反应,反应可在25~30 ℃条件下进行,最高收率可达98%。此外,也有Ni基催化剂的相关报道。2015年,Borzenko等[28]以双(1,5-环辛二烯)镍 (Ni(cod)2) 为催化剂、叔丁醇钠为碱、1, 4-二氧六环/甲苯为溶剂,并使用Josiphos配体 (图5),实现了芳卤与氨的烷基化反应,伯胺的收率为68%~94%。

图4 Cu催化下的Buchwald-Hartwig偶联反应[25-27]
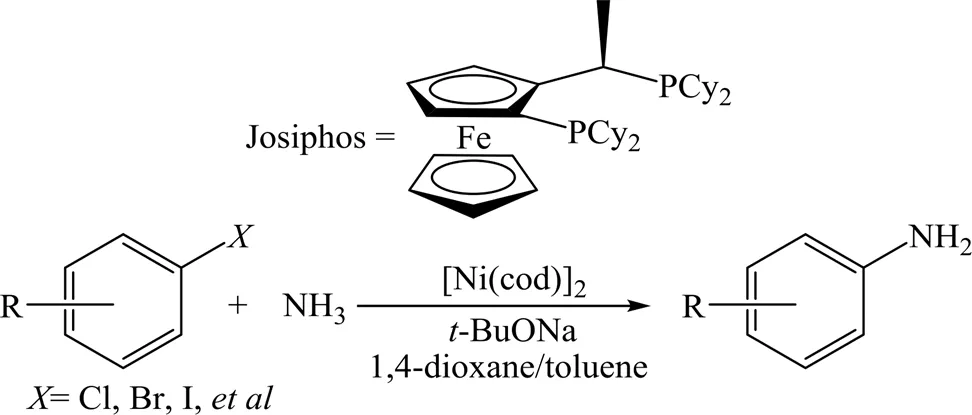
图5 镍催化下以氨为氮源的Buchwald-Hartwig偶联反应[28]
采用Buchwald-Hartwig偶联反应合成伯胺具有步骤短、操作简单的优点,然而该方法只适用于芳基或杂环伯胺的合成。此外,该方法使用均相体系,催化剂回收困难;而且不论是贵金属Pd催化剂,还是非贵金属Cu、Ni催化剂,多数情况下都需添加有机配体,甚至有时需使用二氧杂环等非绿色溶剂。因此,该方法的应用具有局限性。
2.2 烯烃的氢化胺化反应
烯烃作为一种重要的化工原料来源广泛,易于从化石燃料和生物质资源中获取[29]。在烯烃的氢化胺化反应中,通过加成反应的方式直接构筑C─N键,原子经济性或原子效率高达100%,与绿色化学的理念十分相符,烯烃的氢化胺化反应是一条具有现实意义的伯胺化合物合成路线[30]。由于该反应为轻微放热反应且熵变为负值,以及N原子上的孤对电子和富电子的C═C双键存在静电斥力,因此,从热力学和动力学角度看,通过分子间的氢化胺化反应来合成伯胺是一项具有挑战性的工作。
低分子烯烃(C2~C8)可在沸石分子筛催化下和氨反应生成伯胺[31-33],反应通常需在高温(>300 ℃)、高压 (15~30 MPa)下进行,尽管选择性可达90%以上,但转化率较低,一般为十几个百分点。BASF已经通过该方法生产叔丁胺[34](图6)。低分子烯烃还可在NaNH2、CsNH2等碱金属催化剂的作用下[35-36]发生氢化胺化反应,但反应条件苛刻,通常需在81~101 MPa下进行。
2003年,Yasuda等[37]以1,2,4-三苯基苯(1,2,4-TPB)为光敏剂,在二氰基苯(m-DCB)存在条件下,以乙腈/水为溶剂,通过光催化的氢化胺化反应制备了2-氨基-1,2,3,4-四氢萘等伯胺化合物 (图7),然而该方法的底物适用范围较小。
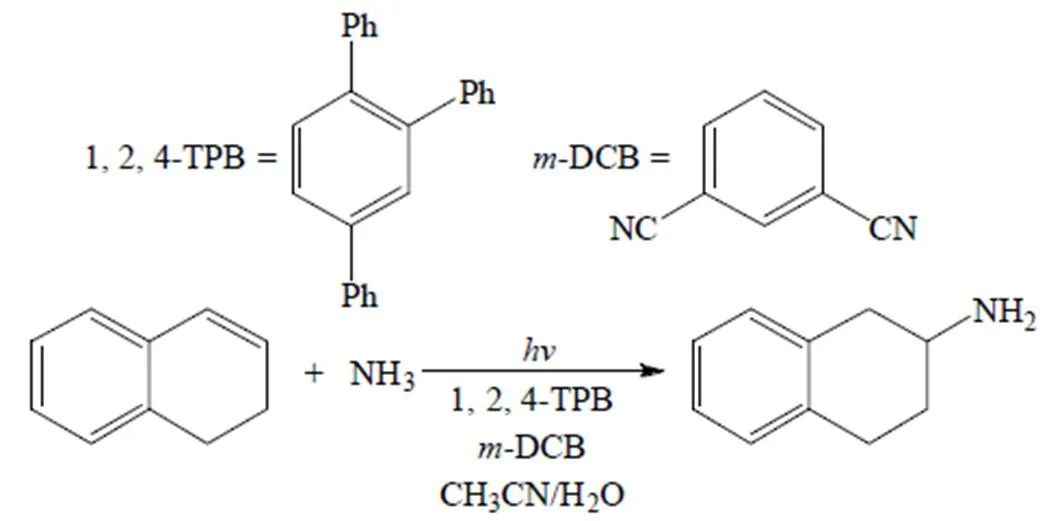
图7 光催化的氢化胺化反应[37]
Zheng等[38]采用一锅法,在温和条件下先使烯烃与Cp2ZrHCl(氢氯二茂锆)加成,然后以2, 4, 6-三甲基苯磺酰羟胺(MSH)为胺化试剂进行胺化反应(图8(a)),目标产品伯胺的收率为62%~88%。2013年,Strom[39]等以羟胺-O-磺酸(HSA)为胺化试剂(图8(b)),在室温条件下考察了该反应,伯胺的收率为71%~94%。然而该类反应的胺化试剂稳定性并不高。
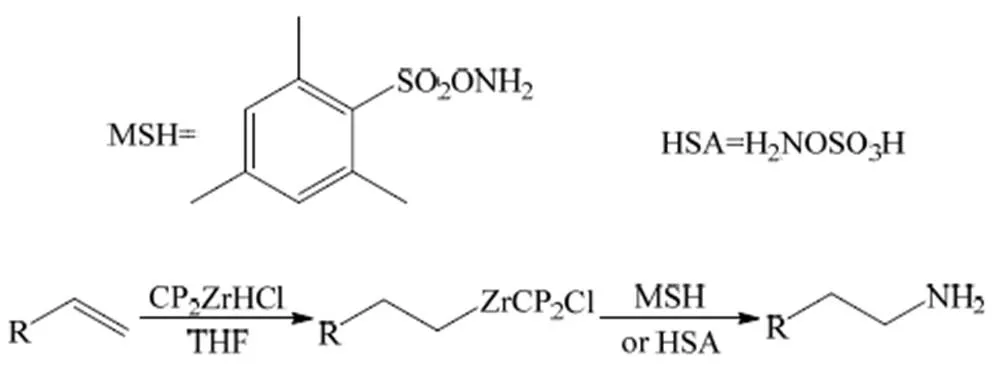
图8 以MSH/HSA为胺化试剂的氢化胺化反应[38-39]
2013年,Miki等[40]和Zhu等[41]报道了CuH配合物催化的烯烃氢化胺化反应。在该反应中,氮原子来源于胺化试剂,氢原子来源于CuH配合物,由于缺乏合适的亲电性胺化试剂,该反应主要用于合成仲胺和叔胺[42]。2018年,Guo等[43]以异恶唑为氮源,以(S)-(+)-5,5’-双[二(3,5-二叔丁基-4-甲氧基苯基)膦]-4,4’-双-1,3-苯并二氧戊环((S)-DTBM SEGPHOS)为配体,以甲基二甲氧基硅烷为氢源,CuH配合物催化的氢化胺化反应,收率为63%~86%。反应机理(图9):CuH配合物首先与烯烃作用得到加成的配合物(I),接着与异恶唑反应生成席夫碱(II),然后与甲基二甲氧基硅烷作用得到中间体(III)和CuH配合物,中间体(III)在盐酸羟胺存在条件下水解得到目标产品伯胺。2019年,Takata等[44]以1-三氟甲基-4-苯基丁烯为原料、N,N-二苄基羟胺为氮源,在CuH配合物催化下进行氢化胺化反应,其中有机膦为配体,聚甲基氢硅氧烷为CuH配合物的氢源。反应首先得到苄基保护的叔胺化合物,然后在Pd催化下脱保护得到目标产品1,1,1-三氟-5-苯基戊烷-2-胺,两步总收率为65%。图中L*为配体。
由于很多有机金属催化剂和强碱性的氨不相容[29],现有的胺化试剂报道较少且适用范围尚待进一步拓展。要实现在温和条件下通过氢化胺化反应构筑C─N键制备伯胺,开发有效的胺化试剂将是突破该反应的关键。

图9 CuH催化的氢化胺化反应机理[43]
2.3 醇的氨解反应
醇化合物来源广泛,工业上可通过烯烃水合、糖发酵以及合成气制备等方式获得,因此,醇氨解制备伯胺是一条具有吸引力的合成路线。此外,醇的氨解反应理论副产物仅为水,还具有原子经济性高、环境友好等优点。低级伯胺化合物通常可通过气相胺化合成,如在350~500 ℃高温下,甲醇和氨气在氧化铝等催化剂作用下于固定床中气相胺化合成甲胺[45]。该方法通常需在高温高压下进行,此外,底物的适用范围也有一定限制。
近年来,随着液相催化反应的发展及“借氢胺化”策略的应用,醇的氨解反应可在更温和的条件下进行[46]。“借氢胺化”是通过质子转移氢化的机理进行的 (图10),醇氧化脱除氢质子生成醛、酮化合物,然后与氨反应生成亚胺,而最初从醇分子上脱除的质子再将亚胺氢化后得到伯胺化合物。因此,术语“借氢”及“质子转移氢化”就是指催化剂“借用”醇分子上的氢,然后“自动”转移到由醛、酮与氨缩合生成的亚胺中间体上。在这个反应中,醇分子不仅作为氢源,还用作底物,理论上不需要外部氢源,相较于其他反应则具有更高的原子经济性。在该反应中,由于生成的伯胺也可与醇分子发生“借氢胺化”反应,因此实际反应产物中会存在仲胺和叔胺等副产物。

图10 借氢胺化反应机理[46]
借氢胺化反应制备伯胺首先采用的是Ru基均相催化剂。2008年,Gunanathan等[47]报道了Ru催化剂RuHCl(A--Pr-PNP)(CO)催化下伯醇的胺化反应,以甲苯为溶剂,在0.76 MPa氨气压力和110 ℃下,目标产物伯胺的最高收率可达96%,反应还可在水相中进行。该催化剂对于仲醇的胺化则不适用。2010年,Imm等[48]和Pingen等[49]以Ru3(CO)12为催化剂、2-(二环己基膦酰基)-1-苯基-1H-吡咯(CataCXium PCy)为配体,分别以2-甲基-2-丁醇和环己烷为溶剂,在较高温度(140~150 ℃)下考察了仲醇的胺化反应,伯胺的最高收率达93%。2011年,Imm等[50]以Ru(CO)ClH(PPh3)3为催化剂,以4, 5-双二苯基膦-9, 9-二甲基氧杂蒽(xantphos)为配体、2-甲基-2-丁醇为溶剂,将该方法的适用范围扩大至二胺类和氨基酯类化合物的合成,目标产物的收率最高可达97%,但反应仍需在较高温度 (130~170 ℃)下进行。尽管均相催化剂活性较高,然而催化剂回收困难,且反应中通常需要添加配体和碱助剂等,从而限制了其发展和应用。利用非均相催化剂可克服这些缺点。有文献报道,负载型的Ru、Pt、Co等催化剂[51-55]可用于醇的胺化反应制备伯胺,然而这些反应通常需要在高压H2氛围下才能进行。因此,从严格意义上说,这类反应本质上应归属于还原胺化反应。最近,Kita等[56]报道了以Ru-MgO/TiO2为催化剂的借氢胺化反应,该催化体系在一系列含吸电子基和供电子基芳香苄醇以及脂肪醇的胺化反应中均表现出良好的反应活性和选择性,以甲苯为溶剂,在0.7 MPa 氨气压力和110 ℃下,伯胺的最高收率达94%。该作者通过亚苄基苯胺与2-羟甲基呋喃的反应产物分析及同位素标记法证明了Ru-MgO/TiO2催化下的胺化反应是按照质子转移氢化的机理进行的;此外,其还利用CO-FTIR、XPS等表征手段证明,MgO可转移电子给Ru,从而抑制Ru-H配合物的分解,进而提高催化剂的反应活性和选择性。
就负载型非贵金属催化剂而言,2013年,Shimizu等[57]报道了Ni/Al2O3催化的仲醇的胺化反应,以邻二甲苯为溶剂,在0.4 MPa 氨气压力和160 ℃下,伯胺收率最高达96%。相比于均相Ru催化剂[48, 50],该催化体系所需氨量更少,并且表现出更高的转换数 (TON)。2014年,Shimizu等[58]又报道了Ni/CaSiO3催化下仲醇的胺化反应,同样取得了较好的反应性能,以邻二甲苯为溶剂,在0.4 MPa 氨气压力和140~170 ℃下,伯胺的收率为70%~88%。然而,Ni催化剂的负载量通常在10%以上,活性组分颗粒易团聚且在反应介质中易浸出,影响催化剂的循环使用性能。2017年,Tomer[59]等以CeO2改性Al2O3,制备了Ni/Ce-Al催化剂,Ni的负载量仅为2%,在以正辛醇为探针分子的借氢胺化反应中表现出较好的活性,以邻二甲苯为溶剂,在0.7 MPa 氨气压力和180 ℃下,正辛胺的收率为60%。
醇的氨解反应具有底物来源广泛、原子经济性高的优点,现有催化剂主要为Ru、Ni催化剂,且多数对醇底物具有区域选择性,开发具有高活性、高选择性且对于醇底物具有较大适用范围的催化剂,尤其是非均相催化剂,将是该方法未来的主要研究任务。
2.4 醛、酮化合物的还原胺化反应
还原胺化反应[60]以醛、酮化合物为底物、氨为氮源、水或醇等为溶剂,在氢化还原剂的作用下反应得到目标产物伯胺,具有工艺简单、原子经济性高的优点,是伯胺类化合物较为实用的合成方法,也是近年来的研究热点。反应可使用HCOOH/HCOONH4(刘卡特-瓦拉赫反应)[61]、NaBH3CN[62]等为氢源,但使用这些还原剂的路线成本较高,并且会导致环境问题。H2作为一种获取简便、清洁高效的氢源,是还原胺化反应的常用还原剂[60]。使用H2的还原胺化反应通常在加压条件下进行,且反应易于控制、收率高,适合大规模工业化生产。
在还原胺化反应中[60](反应机理如图11所示),醛、酮首先与氨发生缩合反应生成亚胺,然后加氢生成伯胺。由于烷基的供电子作用使得生成的伯胺N原子的电子云密度增加,因此具有更高的亲核性,可与醛、酮发生副反应生成亚胺中间体,加氢生成仲胺,仲胺N原子电子云密度又进一步增加,继而再发生副反应生成叔胺。此外,醛、酮还可直接加氢生成醇化合物。虽然调控反应条件参数[63-65],如氨过量、添加铵盐、提高反应温度等都可有效抑制副产物的生成,但是文献认为,伯胺的选择性主要与催化剂的活性金属组分性质有关[60]。
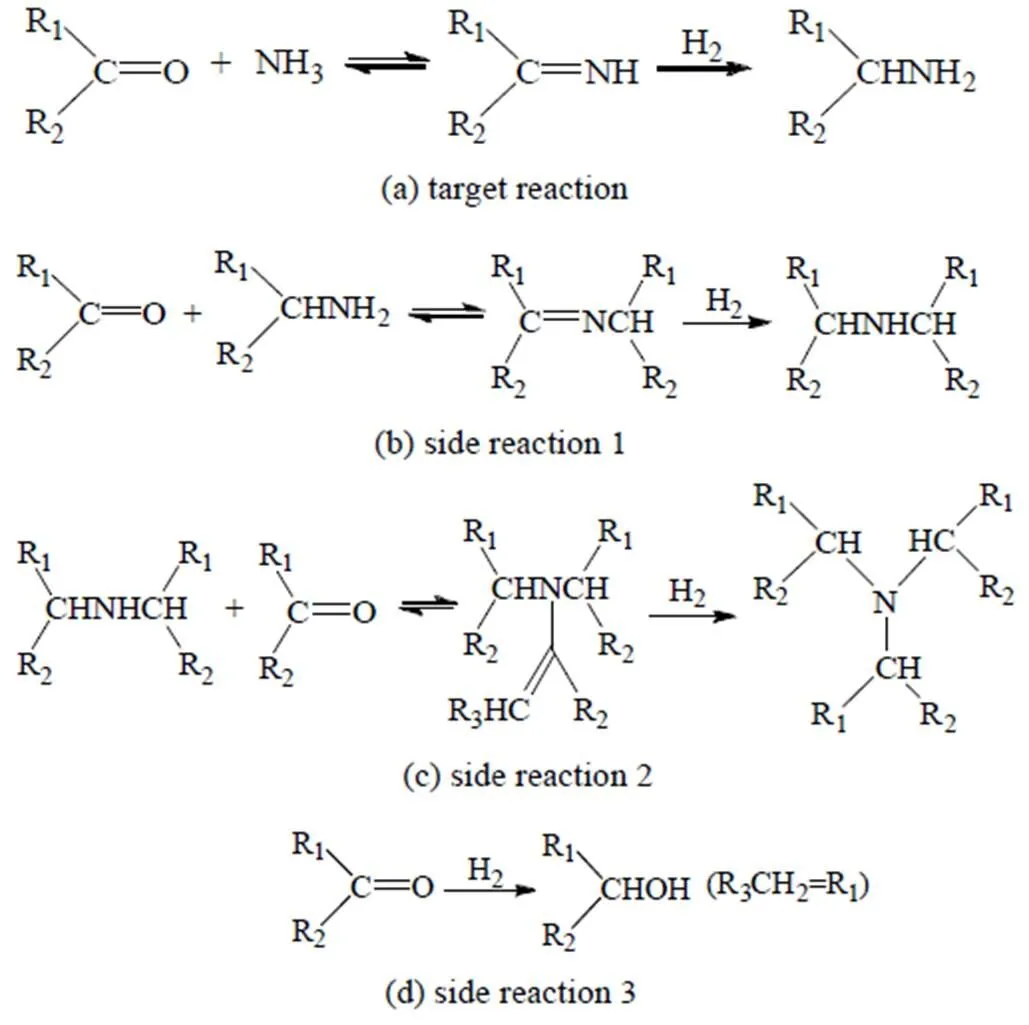
图11 还原胺化反应机理[60]
非均相Ni基催化剂(Raney Ni)是早期使用最多的催化剂[66-68],然而,Raney Ni的使用及反应的后处理比较麻烦,它的活性和选择性相对较低,而且需要在较高温度 ( >100 ℃)和较高压力 ( >10 MPa)下进行催化,耗能大、效率低。近年来,有文献报道[69]使用酸化处理对该催化剂进行优化并用于2, 5-二甲酰基呋喃的还原胺化,但反应仍需在160 ℃这一较高温度下进行,且目标产品2, 5-二(氨基甲基)呋喃的收率小于50%。负载型催化剂及催化剂新型制备技术的发展为Ni基催化剂提供了新的方向,使得反应能够在相对温和的条件下(80~120 ℃、0.1~2 MPa)进行。2019年,Hahn等[70]以邻香兰素、(±)-反式-1, 2-环己二胺和醋酸镍为原料制备了Ni Salen配体 (图12),然后以浸渍法制备了高分散、N掺杂碳嵌入的超顺磁性Ni/Al2O3催化剂。该催化剂对一系列脂肪和芳香醛、酮底物均表现出了良好的活性和选择性,收率通常在80%以上,最高可达99%。此外,该催化剂还适用于药物、生物活性物质以及天然产物的还原胺化反应。Zhang等[71]首先将2, 4-二羟基苯甲酸、六亚甲基四胺、乙二胺与聚醚P123混合、反应得到聚合凝胶,然后通过离子交换和热裂解制备氮掺杂碳负载的镍催化剂MC/Ni,该催化剂在芳香醛的还原胺化反应中表现出较高活性,伯胺的收率为83.2%~99.1%。Murugesan等[72]通过在氩气氛围下热解 (400~1 000 ℃),在二氧化硅上原位生成酒石酸镍络合物,制备了Ni-TA@SiO2催化剂,该催化剂在一系列脂肪和芳香醛、酮底物的还原胺化反应中均表现出了较高的活性,伯胺的收率为84%~96%。Manzoli等[73]制备了Fe3O4@SiO2-Ni核壳催化剂,用于微波辅助的芳香醛的还原胺化反应,在较短时间内转化率可达100%,但Ni的负载量较高,为38.4%。

图12 Ni Salen配体结构图[70]
非均相贵金属(铂族)催化剂近年来常用于还原胺化反应,反应可在相对温和的条件下 (80~100 ℃、0.1~4 MPa)进行,其中以Ru基催化剂报道较多。2002年,Gomez等[64]报道了活性炭负载的Ru催化剂Ru/Cox用于苯甲醛的还原胺化反应,苄胺的收率为77%。2015年,Dong等[74]考察了不同载体负载的Ru催化剂用于线型脂肪醛 (C5~C10) 的还原胺化反应,其中Ru/Al2O3催化剂反应性能最佳,伯胺的收率可达90%以上。2016年,Nishimura等[75]以多元醇还原法制备了Ru-PVP/HAP催化剂 (PVP为聚(N-乙烯基-2-吡咯烷酮)、HAP为羟基磷灰石),并用于糠醛的还原胺化反应,糠胺的收率为60%。2017年,Komanoya等[76]以Ru(NO)(NO3)3为前驱体,通过浸渍法制备了Ru/Nb2O5催化剂并用于芳香以及杂环醛、酮的还原胺化反应,伯胺的最高收率可达99%;催化剂表现出高活性和选择性的原因是活性组分Ru和载体Nb2O5之间的电子转移。Guo等[77]考察了载体Nb2O5形貌对Ru/Nb2O5催化剂活性的影响,层状Nb2O5材料具有最高的表面积,从而产生最高的Ru分散度,因此显示出最高的催化活性。Chandra等[78]通过化学气相沉积法制备了Ru/Ca(NH2)2催化剂,该催化剂的高活性与具有暴露{111}晶面的扁平状Ru纳米微观结构有关。Liang等[79]以浸渍法制备Ru/ZrO2催化剂,通过控制Ru还原深度可得到混合价的表面Ru纳米颗粒,这种微结构同时具有Lewis酸中心和加氢活性中心,使该催化剂在一系列生物质醛、酮的还原胺化反应中表现出较高的活性和选择性。黄龙俊等[80]以环己酮为探针分子,考察了Ru/ZrO2催化剂的反应性能,在研究过程中发现,催化剂的粒径和表面酸性对其活性和选择性有较大影响,在优化条件下,目标产物环己胺的最高收率可达90%。
Pd、Pt、Rh等贵金属催化剂也可用于还原胺化反应[81]。2015年,Nakamura等[82]以浸渍法制备负载型Pt-MoO/TiO2催化剂进行酮化合物的还原胺化,伯胺的收率为60%~77%。2016年,Chatterjee等[83]将商业Rh/Al2O3催化剂用于糠醛的还原胺化反应,糠胺的收率为92%。2004年,Gomez等[84]研究了Pd/C催化的苯甲醛的还原胺化反应,发现经氧化处理的碳载体可提高催化剂活性,并且苄胺的选择性与催化剂中Pd的分散度有关。最近,Jv等[85]以对甲基苄胺为配体制备了Pd-配合物纳米催化剂,反应可在常温常压下进行,伯胺的收率为99%以上。
与非均相贵金属催化体系相比,均相贵金属催化体系所采用的反应温度 (100~135 ℃)和压力 (4.5~6.5 MPa)略高。2002年,Gross等[86]报道了以(Rh(cod)Cl)2((1,5-环辛二烯)氯铑(Ⅰ))二聚体为前驱体、三苯基膦三间磺酸钠盐 (TPPTS)为配体的催化体系,在以苯甲醛为探针分子的还原胺化反应中表现出较高的活性,苄胺的收率为86%。该催化剂还适用于一系列取代的芳香醛底物;对于脂肪醛,则需要使用Rh/Ir双金属催化体系以提高催化性能。2015年,Behr等[63]将该催化体系用于香茅醛的还原胺化反应,伯胺的最高收率为87%。2016年,Gallardo-Donaire等[87]以(PPh)3Ru(CO)ClH(羰基氯氢三(三苯基膦)钌(II))为催化剂,以1,2-双(二苯基膦)乙烷(dppe)为配体、三氟甲烷磺酸盐铝为添加剂,在芳香酮的还原胺化反应中表现出较好的活性,伯胺的收率为56%~99%。2018年,Senthamarai等[88]以RuCl2(PPh3)3(三(三苯基膦)氯化钌(II))为催化剂,在一系列脂肪和芳香醛、酮的还原胺化反应中表现出较高的活性,伯胺的收率为71%~95%。此外,该催化剂对一些类固醇衍生物及药物分子也具有较好的适用性,伯胺的收率为76%~93%。2019年,Murugesan等[89]以Co(BF4)2·6H2O为催化剂,以双(2-二苯基膦乙基)苯基磷(triphos)为配体,在一系列脂肪和芳香醛、酮的还原胺化反应中表现出较高的活性,伯胺的收率为77%~98%。此外,该催化剂对一些生物活性分子也具有较好的适用性,伯胺的收率为78%~92%。
醛、酮化合物的还原胺化法具有工艺绿色、原子经济性高的优点,尤其是对非均相催化体系,由于反应条件温和、收率高且催化剂易于回收利用,从而具有较高的应用价值。然而,石化工业中醛、酮原料的短缺是该方法开发应用的主要瓶颈。由生物质得到醛、酮的方法为还原胺化反应的研究提供了新思路[90]。
3 总结与展望
本文主要总结了卤代化合物与氨的烷基化反应、烯烃的氢化胺化反应、醇的氨解反应和醛、酮化合物的还原胺化反应等通过有机催化反应合成伯胺类化合物的主要方法。
卤代化合物与氨的烷基化反应一般只适用于芳基或杂环伯胺的合成,且该方法使用均相体系、催化剂回收困难;烯烃的氢化胺化反应目前仍然缺乏高效且底物适用范围广的胺化试剂,要在较短时间内实现突破仍存在一定难度。因此,这两种方法都存在一定的局限性,因而限制了其在工业上的应用。
醇的氨解反应和醛、酮化合物的还原胺化反应均以氨为氮源,且理论副产物均为水,具有工艺绿色、环境友好和原子经济性高的优点,从而具有较强的工业化应用价值。在实际过程中,这2种反应都会有仲胺和叔胺等副产物生成,选择具有高活性和高选择性的催化剂则是该两反应的关键。
具体地说,醇的氨解反应,尽管在现有的催化体系下均能以较高收率得到目标产品,但该方法仍有以下两点需要突破。首先,文献报道主要集中在Ru基均相催化剂和Ni基非均相催化剂,因此,需要开发具有高活性、高选择性的新型催化剂,尤其是非均相催化剂,从而拓展该方法的应用范围;此外,现有催化剂多数对醇底物具有区域选择性,因此,需要在反应机理和催化剂的构效关系上进行深入研究,从而开发对醇底物具有较大适用范围的催化体系。
醛、酮的还原胺化反应已有相对成熟的文献报道,铂族贵金属催化剂和Ni、Co等非贵金属催化剂均能催化该反应并且能以较高收率得到目标产品,其中又以非均相催化剂更具优势。研究主要集中在催化剂的新型制备方法,以构筑催化剂特定的微观结构,进而调控其活性和选择性。这些制备方法通常根据金属和载体的特定性质来制定相应的策略,并且特定的反应条件对于催化剂又有特殊的要求。毋庸讳言,在高活性、高选择性新型催化剂的制备方面依然需要投入更多的研究。此外,由于石化醛、酮原料的短缺,生物质醛、酮分子的开发利用将会是该反应的主要研究方向之一。实际上,生物质平台分子的纯化会严重损害过程的经济性,“一锅法”作为“生物质→目标化合物”价值链可持续发展的必要手段又增加了反应的复杂性,对于催化剂也提出了更高的要求。
[1] ROOSE P, ELLER K, HENKES E,. Amines, aliphatic, in Ullmann’s encyclopedia of industrial chemistry [M]. Weinheim: Wiley-VCH, 2015.
[2] TALWAR D, SALGUERO N, ROBERTSON C,. Primary amines by transfer hydrogenative reductive amination of ketones by using cyclometalated IrIIIcatalysts [J]. Chemistry-A European Journal, 2014, 20(1): 245-252.
[3] YANG H, CUI X, DENG Y,. Reductive amination of aldehydes and amines with an efficient Pd/NiO catalyst [J]. Synthetic Communications, 2014, 44(9): 1314-1322.
[4] CHAUDHARI P S, SALIM S D, SAWANT R V,. Sulfated tungstate: A new solid heterogeneous catalyst for amide synthesis [J]. Green Chemistry, 2010, 12(10): 1707-1710.
[5] TAKAHASHI Y, MIYASHI T, YOON U C,. Mechanistic studies of the azomethine ylide-forming photoreactions of N-(silylmethyl)-phthalimides and N-phthaloylglycine [J]. Journal of the American Chemical Society, 1999, 121(16): 3926-3932.
[6] DELEBECQ E, PASCAULT J P, BOUTEVIN B,. On the versatility of urethane/urea bonds: Reversibility, blocked isocyanate, and non-isocyanate polyurethane [J]. Chemical Reviews, 2013, 113(1): 80-118.
[7] HALL H K, BATES R B. Correlation of alkylamine nucleophilicities with their basicities [J]. Tetrahedron Letters, 2012, 53(14): 1830-1832.
[8] VERBICKY J W, WILLIAMS L. Thermolysis of N-alkylsubstituted phthalamic acids. Steric inhibition of imide formation [J]. Journal of Organic Chemistry, 1981, 46(1): 175-177.
[9] 王建红, 李骞, 何丽华, 等. Gabriel合成胺反应的工艺改进 [J]. 化学研究, 2010, 21(4): 48-51.
WANG J H, LI Q, HE L H,. Modification of process for synthesizing polyamine via Gabriel reaction [J]. Chemical Research, 2010, 21(4): 48-51.
[10] 陈程, 罗卓玛, 杨鸿均, 等. 盐酸黄连素的合成研究 [J]. 有机化学, 2016, 36(6): 1426-1430.
CHEN C, LUO Z M, YANG H J,. A novel synthetic route for berberine chloride [J]. Chinese Journal of Organic Chemistry, 2016, 36(6): 1426-1430.
[11] ZAKHARKIN L I, KALININ V N, GEDYMIN V V. The Wolff, Beckmann, Hofmann, Curtius and Schmidt rearrangements in the series of 3-o-carborane derivatives: 1,2-Dicarba-closo-dodecaboranes [J]. Tetrahedron, 1971, 27(6): 1317-1322.
[12] 陈子潇, 沈静, 徐然, 等. 可磁回收Cu-Fe3O4@GE复合材料催化还原对硝基苯酚的研究 [J]. 高校化学工程学报, 2018, 32(3): 577-585.
CHEN Z X, SHEN J, XU R,. Catalytic reduction of-nitrophenol with magnetic Cu-Fe3O4@GE composites [J]. Journal of Chemical Engineering of Chinese Universities, 2018, 32(3): 577-585.
[13] AFAGH N A, YUDIN A K. Chemoselectivity and the curious reactivity preferences of functional groups [J]. Angewandte Chemie International Edition, 2010, 49(2): 262-310.
[14] 邢其毅, 裴伟伟, 徐瑞秋, 等. 基础有机化学[M]. 4版. 北京: 北京大学出版社, 2017.
XING Q Y, PEI W W, XU R Q,.Basic organic chemistry [M]. 4th ed. Beijing: Beijing University Press, 2017.
[15] GURAM A S, BUCHWALD S L. Palladium-catalyzed aromatic aminations with in situ generated aminostannanes [J]. Journal of the American Chemical Society, 1994, 11(17): 7901-7902.
[16] PAUL F, PATT J, HARTWIG J F. Palladium-catalyzed formation of carbon-nitrogen bonds. Reaction intermediates and catalyst improvements in the hetero cross-coupling of aryl halides and tin amides [J]. Journal of the American Chemical Society, 1994, 116(13): 5969-5970.
[17] GRASA G A, VICIU M S, HUANG J,. Amination reactions of aryl halides with nitrogen-containing reagents mediated by palladium/imidazolium salt systems [J]. Journal of Organic Chemistry, 2001, 66(23): 7729-7737.
[18] JAIME-FIGUEROA S, LIU Y, MUCHOWSKI J M,. Allyl amines as ammonia equivalents in the preparation of anilines and heteroarylamines [J]. Tetrahedron Letters, 1998, 39(11): 1313-1316.
[19] SHEN Q, HARTWIG J F. Palladium-catalyzed coupling of ammonia and lithium amide with aryl halides [J]. Journal of the American Chemical Society, 2006, 128(31): 10028-10029.
[20] 梅苏宁, 杨建明, 李亚妮, 等. 钯催化卤代芳烃氨解反应的研究进展 [J]. 化工进展, 2015, 34(10): 3665-3670.
MEI S N, YANG J M, LI Y N,. Progress in palladium-catalyzed ammonolysis of aryl halide [J]. Chemical Industry and Engineering Progress, 2015, 34(10): 3665-3670.
[21] SURRY D S, BUCHWALD S L. Selective palladium-catalyzed arylation of ammonia: Synthesis of anilines as well as symmetrical and unsymmetrical di- and triarylamines [J]. Journal of the American Chemical Society, 2007, 129(34): 10354-10355.
[22] SCHULZ T, TORBORG C, ENTHALER S,. A general palladium-catalyzed amination of aryl halides with ammonia [J]. Chemistry-A European Journal, 2009, 15(18): 4528-4533.
[23] LUNDGREN R J, SAPPONG-KUMANKUMAH A, STRADIOTTO M. A highly versatile catalyst system for the cross-coupling of aryl chlorides and amines [J]. Chemistry-A European Journal, 2010, 16(6): 1983-1991.
[24] VO G D, HARTWIG J F. Palladium-catalyzed coupling of ammonia with aryl chlorides, bromides, iodides, and sulfonates: A general method for the preparation of primary arylamines [J]. Journal of the American Chemical Society, 2009, 131(31): 11049-11061.
[25] KIM J, CHANG S. Ammonium salts as an inexpensive and convenient nitrogen source in the Cu-catalyzed amination of aryl halides at room temperature [J]. Chemical Communications, 2008, (26): 3052-3054.
[26] FANTASIA S, WINDISCH J, SCALONE M. Ligandless copper-catalyzed coupling of heteroaryl bromides with gaseous ammonia [J]. Advanced Synthesis & Catalysis, 2013, 355(4): 627-631.
[27] ABDINE R A A, KURPIK G, WALCZAK A,.Mild temperature amination of aryl iodides and aryl bromides with aqueous ammonia in the presence of CuBr and pyridyldiketone ligands [J]. Journal of Catalysis, 2019, 376: 119-122.
[28] BORZENKO A, ROTTA-LORIA N L, MACQUEEN P M,. Nickel-catalyzed monoarylation of ammonia [J]. Angewandte Chemie International Edition, 2015, 54(12): 3773-3777.
[29] YANG Y, WONG N I, TEO P. Formal intermolecular hydroamination of unbiased olefins for primary amine formation [J]. European Journal of Organic Chemistry, 2015, 2015(6): 1207-1210.
[30] BELLER M, SEAVAD J, TILLACK A,. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends [J]. Angewandte Chemie International Edition, 2004, 43(26): 3368-3398.
[31] PETERSON J O H, FALES H S. Amines via the amination of olefins: US, 4 307 250 [P]. 1981-12-22.
[32] DEEBA M, FORD M E. Heterogeneous acid-catalyzed amination of isobutene to tert-butylamine [J]. Journal of Organic Chemistry, 1988, 53(19): 4594-4596.
[33] MIZUNO N, TABATA M, UEMATSU T,. Amination of 2-methylpropene over proton-exchanged ZSM-5 zeolite catalysts [J]. Journal of Catalysis, 1994, 146(1): 249-256.
[34] CHAUVEL A, DELMON B, HÖLDERICH W F. New catalytic processes developed in Europe during the 1980¢s [J]. Applied Catalysis A: General, 1994, 115(2): 173-217.
[35] HOWK B W, LILLTLE E L, SCOTT S L,. Alkali metal-catalyzed amination of olefins [J]. Journal of the American Chemical Society, 1954, 76(7): 1899-1902.
[36] PEZ G P, GALLE J E. Metal amide catalyzed amination of olefins [J]. Pure and Applied Chemistry, 1985, 57(12): 1917-1926.
[37] YASUDA M, KOJIMA R, TSUTSUI H,. Redox-photosensitized aminations of 1,2-benzo-1,3-cycloalkadienes, arylcyclopropanes, and quadricyclane with ammonia [J]. Journal of Organic Chemistry, 2003, 68(20): 7618-7624.
[38] ZHENG B, SREBNIK M. Amination of zirconocene alkyl chlorides with O-(mesitylsulfonyl) hydroxylamine as a method of preparing primary amines [J]. Journal of Organic Chemistry, 1995, 60(7): 1912-1913.
[39] STROM A E, HARTWIG J F. One-pot anti-Markovnikov hydroamination of unactivated alkenes by hydrozirconation and amination [J]. Journal of Organic Chemistry, 2013, 78(17): 8909-8914.
[40] MIKI Y, HIRANO K, SATOH T,. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines [J]. Angewandte Chemie International Edition, 2013, 52(41): 10830-10834.
[41] ZHU S, NILJIANSKUL N, BUCHWALD S L. Enantio- and regioselective CuH-catalyzed hydroamination of alkenes [J]. Journal of the American Chemical Society, 2013, 135(42): 15746-15749.
[42] PIRNOT M T, WANG Y M, BUCHWALD S L. Copper hydride catalyzed hydroamination of alkenes and alkynes [J]. Angewandte Chemie International Edition, 2016, 55(1): 48-57.
[43] GUO S, YANG J C, BUCHWALD S L. A practical electrophilic nitrogen source for the synthesis of chiral primary amines by copper-catalyzed hydroamination [J]. Journal of the American Chemical Society, 2018, 140(46):15976-15984.
[44] TAKATA T, HIRANO K, MIURA M. Synthesis of‑trifluoromethylamines by Cu-catalyzed regio- and enantioselective hydroamination of 1‑trifluoromethylalkenes [J]. Organic Letters, 2019, 21(11): 4284-4288.
[45] BÄHN S, IMM S, NEUBERT L,. The catalytic amination of alcohols [J]. ChemCatChem, 2011, 3(12): 1853-1864.
[46] SHIMIZU K. Heterogeneous catalysis for the direct synthesis of chemicals by borrowing hydrogen methodology [J]. Catalysis Science & Technology, 2015, 5(3): 1412-1427.
[47] GUNANATHAN C, MILSTEIN D. Synthesis of primary amines directly from alcohols and ammonia [J]. Angewandte Chemie International Edition, 2008, 47(45): 8661-8664.
[48] IMM S, BÄHN S, NEUBERT L,. An efficient and general synthesis of primary amines by ruthenium-catalyzed amination of secondary alcohols with ammonia [J]. Angewandte Chemie International Edition, 2010, 49(44): 8126-8129.
[49] PINGEN D, MÜLLER C, VOGT D. Direct amination of secondary alcohols using ammonia [J]. Angewandte Chemie International Edition, 2010, 49(44): 8130-8133.
[50] IMM S, BÄHN S, ZHANG M,. Improved ruthenium-catalyzed amination of alcohols with ammonia: Synthesis of diamines and amino esters [J]. Angewandte Chemie International Edition, 2011, 50(33): 7599-7603.
[51] PFÜTZENREUTER R, ROSE M. Aqueous-phase amination of biogenic isohexides by using Ru/C as a solid catalyst [J]. ChemCatChem, 2016, 8(1): 251-255.
[52] RUIZ D, AHO A, SALORANTA T,. Direct amination of dodecanol with NH3over heterogeneous catalysts. Catalyst screening and kinetic modelling [J]. Chemical Engineering Journal, 2017, 307: 739-749.
[53] LI Y, CHEN H, ZHANG C,. Reductive amination of 1,6-hexanediol with Ru/Al2O3catalyst in supercritical ammonia [J]. Science China Chemistry, 2017, 60(7): 920-926.
[54] TONG T, GUO W, LIU X,. Dual functions of CoOdecoration in PtCo/CeO2catalysts for the hydrogen-borrowing amination of alcohols to primary amines [J]. Journal of Catalysis, 2019, 378: 392-401.
[55] CHO J H, PARK J H, CHANG T S,. Reductive amination of 2-propanol to monoisopropylamine over Co/-Al2O3catalysts [J]. Applied Catalysis A: General, 2012, 417/418: 313-319.
[56] KITA Y, KUWABARA M, YAMADERA S,. Effects of ruthenium hydride species on primary amine synthesis by direct amination of alcohols over a heterogeneous Ru catalyst [J]. Chemical Science, 2020, 11(36): 9884-9890.
[57] SHIMIZU K, KON K, ONODERA W,. Heterogeneous Ni catalyst for direct synthesis of primary amines from alcohols and ammonia [J]. ACS Catalysis, 2013, 3(1): 112-117.
[58] SHIMIZU K, KANNO S, KON K,. N-alkylation of ammonia and amines with alcohols catalyzed by Ni-loaded CaSiO3[J]. Catalysis Today, 2014, 232: 134-138.
[59] TOMER A, YAN Z, PONCHEL A,. Mixed oxides supported low-nickel formulations for the direct amination of aliphatic alcohols with ammonia [J]. Journal of Catalysis, 2017, 356: 133-146.
[60] GOMEZ S, PETERS J A, MASCHMEYER T. The reductive amination of aldehydes and ketones and the hydrogenation of nitriles: Mechanistic aspects and selectivity control [J]. Advanced Synthesis & Catalysis, 2002, 344(10): 1037-1057.
[61] KADYROV R, RIERMEIER T H. Highly enantioselective hydrogen-transfer reductive amination: Catalytic asymmetric synthesis of primary amines [J]. Angewandte Chemie International Edition, 2003, 42(44): 5472-5474.
[62] ASHWEEK N J, COLDHAM I, VENNALL G P. A convenient route to N-alkyl-2-tributylstannyl-pyrrolidines involving reductive amination [J]. Tetrahedron Letters, 2000, 41(13): 2235-2237.
[63] BEHR A, WINTZER A, LÜBKE C,. Synthesis of primary amines from the renewable compound citronellal via biphasic reductive amination [J]. Journal of Molecular Catalysis A: Chemical, 2015, 404/405: 74-82.
[64] GOMEZ S, PETERS J A, VAN DER WAAL J C,. Preparation of benzylamine by highly selective reductive amination of benzaldehyde over Ru on an acidic activated carbon support as the catalyst [J]. Catalysis Letters, 2002, 84(1/2): 1-5.
[65] GOMEZ S, PETERS J A, VAN DER WAAL J C,. High-throughput experimentation as a tool in catalyst design for the reductive amination of benzaldehyde [J]. Applied Catalysis A: General, 2003, 254(1): 77-84.
[66] WINANS C F, ADKINS H. The preparation of amines by catalytic hydrogenation of derivatives of aldehydes and ketones [J]. Journal of the American Chemical Society, 1933, 55(5): 2051-2058.
[67] SCHWOEGLER E J, ADKINS H. Preparation of certain amines [J]. Journal of the American Chemical Society, 1939, 61(12): 3499-3502.
[68] DOLEŽAL P, MACHALICKÝ O, PAVELEK M,. Reductive amination of cyclopentanone [J]. Applied Catalysis A: General, 2005, 286(2): 202-210.
[69] LE N T, BYUN A, HAN Y,. Preparation of 2,5-bis(aminomethyl)furan by direct reductive amination of 2,5-diformylfuran over Nickel-Raney catalysts [J]. Green and Sustainable Chemistry, 2015, 5(3): 115-127.
[70] HAHN G, KUNNAS P, DE JONGE N,. General synthesis of primary amines via reductive amination employing a reusable nickel catalyst [J]. Nature Catalysis, 2019, 2(1): 71-77.
[71] ZHANG Y, YANG H, CHI Q,. Nitrogen-doped carbon-supported nickel nanoparticles: A robust catalyst to bridge the hydrogenation of nitriles and the reductive amination of carbonyl compounds for the synthesis of primary amines [J]. ChemSusChem, 2019, 12(6): 1246-1255.
[72] MURUGESAN K, BELLER M, JAGADEESH R V. Reusable nickel nanoparticles-catalyzed reductive amination for selective synthesis of primary amines [J]. Angewandte Chemie International Edition, 2019, 58(15): 5064-5068.
[73] MANZOLI M, GAUDINO E C, CRAVOTTO G,. Microwave-assisted reductive amination with aqueous ammonia: Sustainable pathway using recyclable magnetic nickel-based nanocatalyst [J]. ACS Sustainable Chemistry & Engineering, 2019, 7(6): 5963-5974.
[74] DONG B, GUO X, ZHANG B,. Heterogeneous Ru-based catalysts for one-pot synthesis of primary amines from aldehydes and ammonia [J]. Catalysts, 2015, 5(4): 2258-2270.
[75] NISHIMURA S, MIZUHORI K, EBITANI K. Reductive amination of furfural toward furfurylamine with aqueous ammonia under hydrogen over Ru-supported catalyst [J]. Research on Chemical Intermediates, 2016, 42(1): 19-30.
[76] KOMANOYA T, KINEMURA T, KITA Y,. Electronic effect of ruthenium nanoparticles on efficient reductive amination of carbonyl compounds [J]. Journal of the American Chemical Society, 2017, 139(33): 11493-11499.
[77] GUO W, TONG T, LIU X,. Morphology-tuned activity of Ru/Nb2O5catalyst for ketone reductive amination [J]. ChemCatChem, 2019, 11(16): 4130-4138.
[78] CHANDRA D, INOUE Y, SASASE M,. A high performance catalyst of shape-specific ruthenium nanoparticles for production of primary amines by reductive amination of carbonyl compounds [J]. Chemical Science, 2018, 9(27): 5949-5956.
[79] LIANG G, WANG A, LI L,. Production of primary amines by reductive amination of biomass-derived aldehydes/ketones [J]. Angewandte Chemie International Edition, 2017, 56(11): 3050-3054.
[80] 黄龙俊, 鲍佳浩, 郭璐瑶, 等. Ru/ZrO2催化环己酮还原胺化制备环己胺工艺 [J]. 化学反应工程与工艺, 2019, 35(3): 274-281.
HUANG L J, BAO J H, GUO L Y,. Study on Ru/ZrO2catalyzed reductive amination of cyclohexanone to cyclohexylamine [J]. Chemical Reaction Engineering and Technology, 2019, 35(3): 274-281.
[81] BÓDIS J, LEFFERTSD L, MÜLLER T E,. Activity and selectivity control in reductive amination of butyraldehyde over noble metal catalysts [J]. Catalysis Letters, 2005, 104(1/2): 23-28.
[82] NAKAMURA Y, KON K, TOUCHY A S,. Selective synthesis of primary amines by reductive amination of ketones with ammonia over supported Pt catalysts [J]. ChemCatChem, 2015, 7(6): 921-924.
[83] CHATTERJEE M, ISHIZAKA T, KAWANAMI H. Reductive amination of furfural to furfurylamine using aqueous ammonia solution and molecular hydrogen: An environmentally friendly approach [J]. Green Chemistry, 2016, 18(2): 487-496.
[84] GOMEZ S, PETERS J A, VAN DER WAAL J C,. The rationalization of catalyst behaviour in the reductive amination of benzaldehyde with ammonia using a simple computer model [J]. Applied Catalysis A: General, 2004, 261(1): 119-125.
[85] JV X, SUN S, ZHANG Q,. Efficient and mild reductive amination of carbonyl compounds catalyzed by dual-function palladium nanoparticles [J]. ACS Sustainable Chemistry & Engineering, 2020, 8(3): 1618-1626.
[86] GROSS T, SEAYAD A M, AHMAD M,. Synthesis of primary amines: first homogeneously catalyzed reductive amination with ammonia [J]. Organic Letters, 2002, 4(12): 2055-2058.
[87] GALLARDO-DONAIRE J, ERNST M, TRAPP O,. Direct synthesis of primary amines via ruthenium-catalysed amination of ketones with ammonia and hydrogen [J]. Advanced Synthesis & Catalysis, 2016, 358(3): 358-363.
[88] SENTHAMARAI T, MURUGESAN K, SCHNEIDEWIND J,. Simple ruthenium-catalyzed reductive amination enables the synthesis of a broad range of primary amines [J]. Nature Communications, 2018, 9(8): 4123-4134.
[89] MURUGESAN K, WEI Z, CHANDRASHEKHAR V G,. Homogeneous cobalt-catalyzed reductive amination for synthesis of functionalized primary amines [J]. Nature Communications, 2019, 10(11): 5443-5451.
[90] PELCKMANS M, RENDERS T, VAN DE VYVER S,. Bio-based amines through sustainable heterogeneous catalysis [J]. Green Chemistry, 2017, 19(22): 5303-5331.
Progress in synthesis of primary amine compounds
YU Jie1,2, LONG Yi-hua1, LI Wang-tao1, WANG Zheng-bao1
(1. College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China;2. College of Biology and Environmental Engineering, Zhejiang Shuren University, Hangzhou 310015, China)
Primary amines are a kind of important organic chemical raw materials, and are widely used in medicine, pesticide, food additive, detergent, lubricant and functional polymer materials. The paper mainly summarized the main methods of synthesizing primary amines through catalytic reactions, such as alkylation of halogenated compounds with ammonia, hydroamination of olefins, ammonolysis of alcohols and reductive amination of aldehydes and ketones. The catalytic systems used in each method were emphatically reviewed, and the related reaction mechanisms were briefly introduced. The ammonolysis of alcohols and the reductive amination of aldehydes and ketones are two methods with strong industrial prospects. The main development direction of these two methods in the future is to develop new catalytic systems and their preparation methods to effectively improve the activity, selectivity and application scope of catalysts.
primary amine; synthesis; catalysis; ammonolysis; reductive amination
1003-9015(2021)06-0955-11
O643.38
A
10.3969/j.issn.1003-9015.2021.06.002
2021-03-03;
2021-06-01。
国家自然科学基金(21676238);浙江省公益技术研究计划(LGJ20B060002);浙江树人大学省属高校基本科研业务费专项资金(2021XZ016)。
俞杰(1984-),男,浙江杭州人,浙江大学博士后,博士。
王正宝,E-mail:zbwang@zju.edu.cn
猜你喜欢
中学生数理化·中考版(2021年12期)2021-12-31
能源化工(2021年2期)2021-12-30
中学生数理化·中考版(2021年11期)2021-12-06
中学化学(2017年6期)2017-10-16
材料科学与工程学报(2016年4期)2017-01-15
广东石油化工学院学报(2016年6期)2016-05-17
合成化学(2015年4期)2016-01-17
合成化学(2015年5期)2015-03-26
石油化工应用(2014年2期)2014-03-11
郑州大学学报(理学版)(2014年2期)2014-03-01
